Publié le 30 aoû 2010Lecture 14 min
Panorama des génodermatoses avec atteinte pilaire
O. DEREURE, Service de dermatologie, CHU de Montpellier

Un certain nombre d’affections héréditaires se manifestant exclusivement, essentiellement ou en partie par des anomalies des cheveux sont désormais bien connues et ont été décryptées sur le plan biochimique. Les dermatologues sont bien entendu en première ligne pour le diagnostic et la prise en charge de ces entités et se doivent de connaître les rudiments de ces syndromes parfois complexes, qui peuvent dépasser le strict cadre cutané.
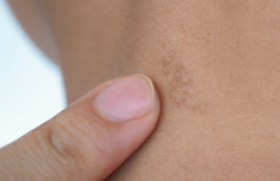
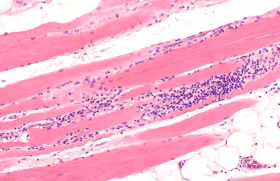
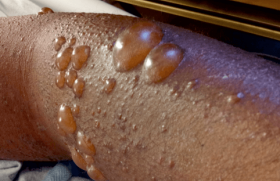
On peut classer les génodermatoses avec anomalies des cheveux en : – affections qualitatives et/ou quantitatives isolées de la tige pilaire, sans modification cutanée ou extracutanée associée ; – affections touchant principalement les cheveux et la peau, qu’il s’agisse du cuir chevelu ou de zones non pileuses ; – affections touchant principalement d’autres structures avec atteinte connexe, mais au deuxième plan des cheveux et selon les cas des téguments. Génodermatoses avec atteintes isolées de la tige pilaire Il s’agit des dysplasies pilaires au sens large du terme et des hypotrichoses sans atteinte du scalp. Dysplasies pilaires isolées Elles ne sont pas toutes d’origine génétique au sens exact du terme, mais nombre d’entre elles évoluent quand même dans un contexte familial même si le mode de transmission n’est pas bien défini. Dans d’autres cas, elles sont sporadiques et ne correspondent donc pas à des génodermatoses « vraies ». Formes alopéciantes • La trichorrhexie noueuse est la dysplasie pilaire la plus fréquente, et le plus souvent isolée. Elle peut être héréditaire et alors souvent congénitale, de transmission autosomique dominante ou acquise par agressions mécaniques, physiques ou chimiques répétées des cheveux. L’anomalie morphologique est liée à une fragilisation des cellules de la cuticule, qui devient discontinue, entraînant une accumulation de cellules du cortex formant une sorte de noeud, chaque nodosité étant une zone de fragilité pouvant aboutir à des fractures des tiges pilaires. La trichorrhexie noueuse peut aussi être un des éléments de maladies génétiques plus complexes telles que l’acidurie argininosuccinique, le syndrome Kabuki, ou encore le syndrome de Bazex-Dupré-Christol. • Les pili torti représentent un groupe complexe d’affections où les anomalies sont limitées aux cheveux ou plus diffuses. Cette dysplasie pilaire est assez ubiquitaire et peut se rencontrer dans des situations très diverses, d’origine génétique ou non. Les cheveux sont aplatis avec des torsions de 90 à 360° tous les 0,5 à 1 mm (figure 1), apparaissent cassants, raréfiés et ont souvent un aspect ébouriffé et incoiffable. Cette dystrophie peut être isolée, congénitale ou d’apparition plus tardive, en général de transmission autosomique dominante quand elle est familiale. Figure 1. Pili torti (maladie de Menkès) (coll. Dr C. Labrèze, CHRU Bordeaux). Divers syndromes peuvent s’associer à des pili torti, notamment le syndrome de Menkès, des affections avec déficit neurosensoriel ainsi que les syndromes de Rapp-Hodgkin et de Bazex-Dupré-Christol. • Une forme particulière de cheveux laineux diffus associés à une hypotrichose, de transmission autosomique récessive, a été récemment rapportée à des mutations du gène P2RY5, qui code pour un récepteur « orphelin » (c’est-à-dire dont le ligand physiologique est encore inconnu) couplé à une protéine G. Les mutations sont souvent ponctuelles, faux-sens. Formes non alopéciantes Elles sont très diverses : • pili annulati avec une succession de bandes sombres normales et claires (présence d’air dans la corticale) avec un aspect global de « bâton de Mikado » ; • syndrome des cheveux incoiffables diffus ou localisés de transmission probablement autosomique dominante, lié à une anomalie structurale de la tige pilaire, qui prend une section triangulaire ou réniforme (pili trianguli ou canaliculi). Un aspect de type pili canaliculi peut plus rarement être observé dans des affections alopéciantes telles que l’hypotrichose de type Marie Unna, certaines dysplasies ectodermiques (syndrome de Rapp- Hodgkin), le syndrome des cheveux anagènes caduques ; • trichoptylose : fractures longitudinales de la tige donnant un aspect de cheveux « fourchus » ; • cheveux laineux isolés ou wooly hair : cette dysplasie est liée à une torsion importante de la tige pilaire, qui a souvent une section elliptique. L’anomalie peut être localisée, sous la forme d’un hamartome à cheveux laineux, ou atteindre l’ensemble du scalp dans une forme autosomique dominante ou récessive beaucoup plus rare. Les formes autosomiques récessives peuvent également être syndromiques quand elles s’associent à une kératodermie palmo-plantaire (cf. infra). Hypotrichoses isolées ou avec atteinte mineure des téguments Hypotrichose simplex du scalp Cette variété d’hypotrichose qui atteint exclusivement le scalp est probablement liée à des mutations non-sens du gène codant pour la cornéodesmosine, glycoprotéine exprimée dans l’épiderme et dans la tige pilaire interne et qui a un rôle de molécule d’adhésion interkératinocytaire. Hypotrichose de Marie Unna Cette affection autosomique dominante rare est caractérisée par l’apparition d’une alopécie de type androgénique durant l’enfance avec des anomalies de la tige pilaire, qui est aplatie et épaissie, souvent tordue (figure 2/illustration) ; elle est peut-être liée à des mutations d’une région inhibitrice située en amont du gène hairless mis en cause dans l’alopécie congénitale universelle et atrichie avec lésions papuleuses (cf. infra). Syndrome des cheveux anagènes caduques Ce syndrome, peut-être plus fréquent qu’initialement estimé, est le plus souvent sporadique et acquis, mais un certain nombre de cas familiaux ont été rapportés avec une transmission plutôt autosomique dominante. Il se caractérise essentiellement par l’apparition progressive chez l’enfant, parfois plus brutale chez l’adolescent ou l’adulte, d’anomalies pilaires avec arrachage facile et sans douleur de touffes de cheveux, souvent par l’enfant luimême, associée à une croissance lente des cheveux qui restent courts, aboutissant à une alopécie progressive et diffuse ou au contraire en plaques. Le trichogramme est tout à fait caractéristique, constitué presque exclusivement de cheveux en phase anagène (80 % à 100 %), dystrophiques pour au moins 50 % d’entre eux, dépourvus de gaine épithéliale ou munis d’une gaine épithéliale retroussée prenant un aspect en « chaussettes descendues » (cette anomalie n’est pas spécifique). Les racines sont souvent tordues et rétrécies, et prennent souvent un aspect en « queue de souris » ou en hameçon avec absence de zone kératogène. La physiopathologie de ce syndrome est mal connue. Alopécie congénitale universelle Cette affection sera traitée plus bas en même temps que l’atrichie avec lésions papuleuses puisque ces deux entités sont alléliques. Hypotrichose localisée autosomique récessive Cette alopécie héréditaire de description récente affecte principalement le scalp, les sourcils, les cils, le tronc et les extrémités, mais épargne largement la pilosité faciale, pubienne et axillaire. Les cheveux et poils atteints sont fragiles et se cassent facilement en laissant des cheveux résiduels épars et courts d’aspect assez caractéristique. Figure 3. Monilethrix : aspect moniliforme des cheveux (coll. Dr C. Labrèze, CHRU Bordeaux). Il s’y associe souvent une hyperkératose folliculaire donnant un aspect « rugueux », un érythème et un état squameux et prurigineux prédominant sur l’extrémité céphalique. Cette génodermatose est liée à des anomalies (délétion homozygote) d’un gène codant pour la desmogléine 4. Génodermatoses avec atteinte élective ou principale des cheveux et des téguments Monilethrix Le monilethrix est une dystrophie capillaire héréditaire rare qui se transmet le plus souvent sur un mode autosomique dominant, et qui se caractérise par une kératose pilaire souvent discrètement inflammatoire et prédominante sur la région occipitale, une raréfaction progressive des cheveux, là encore surtout postérieure et un aspect en chapelet, moniliforme, des tiges pilaires, qui sont souvent fragiles (figures 3 et 4). Dans les formes autosomiques dominantes, diverses mutations ont été retrouvées sur des gènes codant pour des kératines pilaires (hHb1, hHb3 et hHb6) mutations portant notamment des résidus hautement conservés et qui entraînent très probablement des modifications structurelles importantes de la molécule. Le phénotype pathologique est parfois très variable d’une famille à une autre ; l’intervention de facteurs non génétiques pourrait être à l’origine de cette variabilité phénotypique. Figure 4. Monilethrix : alopécie à prédominance occipitale (coll. Pr J. Mazereeuw, CHRU Toulouse). Une forme autosomique récessive a été plus rarement décrite, liée à des mutations ponctuelles de la desmogléine 4 donnant lieu à une forme de chevauchement entre monilethrix et hypotrichose autosomique récessive localisée. Syndrome de Netherton Le syndrome de Netherton est une génodermatose autosomique récessive caractérisée par une érythrodermie congénitale, des manifestations de type atopique avec dermite atopique et rhinite, une hyperéosinophilie, une élévation des IgE totales et des anomalies tout à fait spécifiques de la tige pilaire à type de Trichorrhexis invaginita ou cheveux « bambous » par invagination de la partie distale de la tige pilaire dans sa partie proximale (figures 5 et 6). Les lésions cutanées évoluent ensuite vers un érythème squameux souvent caractérisé par une double bordure, parfois mobile en fonction du temps (érythème circonflexe linéaire de Gomel). Il peut s’y associer des infections, un retard de croissance et des épisodes de déshydratation avec un risque vital non négligeable. Cette génodermatose sévère est liée à des mutations de spink 5, qui code pour une protéine à activité inhibitrice des sérines protéases telles les kallikréines épidermiques, la protéine LEKTI pour Lympho-Epithelial Kazal-Type-related Inhibitor et il est probable que les mutations en cause soient directement à l’origine du phénotype pathologique. Un espoir thérapeutique est représenté par l’emploi d’inhibiteurs de protéases topiques. Alopécie congénitale universelle et atrichie avec lésions papuleuses Ces deux entités autosomiques récessives très proches, qui sont peut-être moins rares qu’on ne le pense généralement et pourraient constituer un diagnostic différentiel des pelades décalvantes précoces, sont liées, dans la majorité des cas, à des mutations d’un même gène (hairless), mais d’autres gènes pourraient être en cause (récepteur à la vitamine D, ornithine décarboxylase ou récepteur RXR alpha à l’acide 9-cis rétinoïque). Le tableau clinique de l’alopécie universelle congénitale est caractérisé par la présence de cheveux épars à la naissance, cheveux qui disparaissent complètement dans un délai de quelques mois avec, en général, disparition conjointe des cils et des sourcils. Figure 5. Syndrome de Netherton : érythrodermie néonatale (coll. Dr C. Labrèze, CHRU Bordeaux). Dans l’atrichie avec lésions papuleuses, la chute pilaire est tout à fait similaire, mais il s’y associe le développement progressif de papules folliculaires multiples, sur le cuir chevelu, mais aussi sur d’autres zones cutanées, papules qui correspondent à des kystes épithéliaux. Syndrome de Bazex-Dupré-Christol et entités apparentées Le syndrome de Bazex-Dupré- Christol est une génodermatose rare, transmise sur un mode dominant lié à l’X et qui apparaît dès la période néonatale ou dans la petite enfance. Ses manifestations cliniques associent atrophodermie folliculaire prédominant sur le dos des mains et des pieds et le visage, hypotrichose du cuir chevelu et parfois des sourcils, hypohidrose, grains de milium et survenue précoce de carcinomes basocellulaires au cours de la deuxième ou de la troisième décennie. Le gène impliqué a été localisé sur le bras long du chromosome X en position Xq24- q27.1, mais l’implication du gène UBE2A n’a pas été confirmée pour l’instant. Les principaux diagnostics différentiels sont probablement la nævomatose basocellulaire et la chondrodysplasie ponctuée dominante liée à l’X, où une atrophodermie folliculaire est également présente. Syndromes avec cheveux laineux et kératodermie palmo-plantaire Plusieurs syndromes plus ou moins bien individualisés associant cheveux laineux, kératodermie palmo-plantaire et cardiomyopathie droite ou gauche, ont été rapportés à des mutations de gènes codant pour des protéines de la plaque desmosomale, notamment : • le syndrome de Naxos, génodermatose autosomique récessive rare avec cardiomyopathie ventriculaire droite génératrice de troubles du rythme, kératodermie palmo-plantaire non épidermolytique diffuse et cheveux laineux (mutations du gène codant pour la plakoglobine) ; Figure 6. Syndrome de Netherton : cheveux « bambous » (coll. Dr C. Labrèze, CHRU Bordeaux). • et le syndrome de Carvajal autosomique récessif avec kératodermie striée généralisée précoce, affectant particulièrement la région palmo-plantaire, cheveux laineux et cardiomyopathie ventriculaire gauche dilatée avec risque de mort prématurée par insuffisance cardiaque (mutations du gène codant pour la desmoplakine). Génodermatoses avec atteinte non dominante des cheveux et des téguments Dans ces affections, les anomalies des cheveux et des téguments existent, mais ne sont pas forcément au premier plan du tableau clinique. Trichothiodystrophie de Vera Price Ce terme regroupe en fait différentes entités autosomiques récessives dont le point commun est une dysplasie pilaire très particulière, avec des cheveux secs, rares, fragiles, pauvres en soufre et cystéine, anormaux morphologiquement (fractures transverses, alternance de bandes sombres et claires en lumière polarisée avec un aspect « tigré » et une cuticule anormale ou absente). Cette dysplasie rare résulte d’une diminution de l’expression des protéines riches en soufre, qui sont aussi qualitativement altérées et anormalement distribuées dans le cortex et la cuticule pilaire. La moitié environ des patients atteints de trichothiodystrophie présentent également une photosensibilité qui peut s’intégrer dans un syndrome PIBIDS (Photosensibilité, Ichtyosis, Brittle hair, Impaired intelligence, Decreased fertility and Short stature). Cette photosensibilité se traduit sur le plan clinique, mais également et surtout sur le plan moléculaire. Dans la moitié des cas, il existe une anomalie de la réparation des lésions UV-induites de l’ADN impliquant le système d’excisionréparation, ce qui rapproche la trichothiodystrophie du syndrome de Cockayne et de Xeroderma pigmentosum, où ce système est également déficitaire. Sur le plan génétique, trois groupes de complémentation (TTD-A, TTD-B [le plus fréquent] et TTD-C) ont été individualisés dans les trichothiodystrophies avec photosensibilité et anomalies du système d’excision-réparation des lésions UV-induites de l’ADN, aussi observées dans le syndrome de Cockayne et dans le Xeroderma pigmentosum. D’autres gènes impliqués sont également mutés dans le syndrome de Cockayne et/ou dans le Xeroderma pigmentosum, mais avec des profils de mutation différents, ce qui explique sans doute l’absence d’évolution tumorale. Maladie de Menkès Cette affection récessive et liée à l’X, très rare (un nouveau-né mâle sur 200 000), est redoutable en raison des multiples anomalies viscérales qui caractérisent ce syndrome, notamment vasculaires avec anévrysmes, osseuses et neurologiques. Les anomalies pilaires, mais également viscérales sont sous-tendues par un mécanisme commun de déficience des enzymes fonctionnant avec du cuivre, notamment la cytochrome C oxydase, la lysil-oxydase et une oxydase pour l’instant non caractérisée qui catalyse la formation de ponts entre les molécules de kératine. Cette déficience relative en cuivre est elle-même liée à une anomalie d’un gène codant pour une protéine de transport intracytoplasmique du cuivre qui a une activité ATPase, l’ATP7A. Les mutations en cause sont nombreuses avec une sévérité particulière des mutations non-sens aboutissant à une protéine tronquée dont la fonction de transport est quasi nulle. Dysplasies ectodermiques Ces syndromes très divers se transmettent en général sur un mode autosomique, plutôt récessif que dominant, et associent des altérations des phanères (cheveux, ongles), des dérivés épidermiques telles les glandes sudorales, des dents et des anomalies cutanées ou extracutanées très diverses. Leur classification est très complexe (29) et les exemples les plus connus sont : la dysplasie ectodermique anhydrotique ou syndrome de Christ-Siemens- Touraine lié à l’X ; le syndrome AEC, associant ankyloblépharon, dysplasie ectodermique et fente labio-palatine ; le syndrome EEC, associant ectrodactylie, dysplasie ectodermique et fente labio-palatine ; et le syndrome de Rapp- Hodgkin, ces deux affections étant liées à des mutations du facteur de transcription nucléaire p63. Des mutations de la plakophiline 1 peuvent entraîner une variété particulière de dysplasie ectodermique, avec fragilité cutanée et des cheveux, anomalies unguéales, et hypohidrose. Syndromes avec vieillissement prématuré La plupart des syndromes génétiques avec vieillissement prématuré systémique s’accompagnent de modifications des cheveux avec hypotrichose, voire alopécie et canitie précoces : progeria de Hutchinson-Gilford, syndrome de Werner, syndrome de Cockayne. En revanche, le syndrome de Bloom ne modifie pas, en principe, l’aspect et la densité des cheveux, tandis que le syndrome de Rothmund-Thomson et l’ataxietélangiectasie peuvent s’accompagner d’un grisonnement prématuré des cheveux. Anomalies du métabolisme des acides aminés Certaines affections génétiques impliquant des troubles du métabolisme des acides aminés peuvent entraîner des anomalies pigmentaires des cheveux, notamment une hypopigmentation (métabolisme de l’histidine, de la phénylalanine, etc.) mais aussi, comme signalé ci-dessus, une hypotrichose avec dysplasie pilaire (acidurie argininosuccinique, citrulinémie, etc.).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :