Publié le 26 nov 2009Lecture 12 min
Lymphomes T cutanés : les « bons » et les « mauvais »
S. INGEN-HOUSZ-ORO, M. BAGOT Service de dermatologie, CHU Henri Mondor, Créteil
Les lymphomes T cutanés (LTC) sont des lymphoproliférations T qui touchent exclusivement la peau au moment du diagnostic. Ceci les différencie des lymphomes T primitivement extracutanés à extension cutanée secondaire. La distinction entre LTC et lymphome cutané secondaire est importante sur le plan pronostique et thérapeutique.
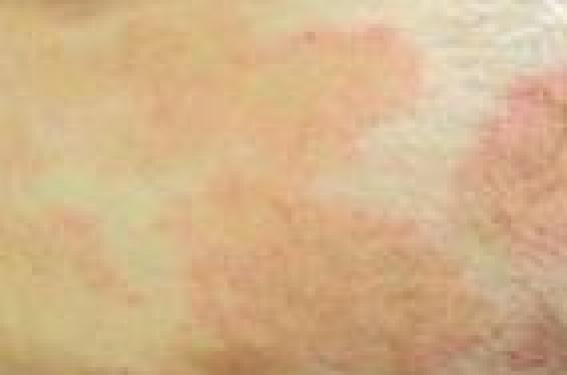
Le pronostic des lymphomes T cutanés (LTC) dépend essentiellement du type histologique et de l’extension de la maladie. La classification histo-pronostique OMS-EORTC (1) permet de distinguer, d’une part, les LTC indolents, comme le mycosis fongoïde et ses variantes ou les lymphoproliférations CD30+, et d’autre part, les LTC agressifs comme le syndrome de Sézary ou les lymphomes CD30- (tableau 1). Le bilan d’extension permet de définir le stade de la maladie selon la classification TNM (2,3). Les choix thérapeutiques dépendent donc du type anatomo-clinique, et pour un même type de lymphome, du bilan d’extension. Le mycosis fongoïde et le syndrome de Sézary Mycosis fongoïde Lymphome T cutané le plus fréquent, le mycosis fongoïde (MF) est caractérisé par son évolution très lente sur des années et son bon pronostic. Il se présente sous forme de plaques initialement non infiltrées, de taille variable, à limites nettes volontiers encochées, touchant une surface corporelle plus ou moins étendue (figure 1), évoluant très lentement vers des plaques infiltrées et parfois des tumeurs, voire une érythrodermie. Le prurit est variable. Certaines formes cliniques sont trompeuses : MF hyper- ou hypopigmenté, MF psoriasiforme, verruqueux, bulleux… Le MF pilotrope ou folliculotrope est caractérisé par le tropisme pilaire des cellules tumorales, associé ou non à une mucinose folliculaire. La profondeur de l’infiltrat rend cette forme de MF moins sensible aux traitements locaux et grève un peu son pronostic. Cliniquement, des plaques (papules ou papulo-nodules) avec ou sans alopécie, prédominant sur l’extrémité céphalique, sont évocatrices. Le MF pagétoïde ou maladie de Woringer- Kolopp, de très bon pronostic, est caractérisé par une lésion annulaire unique souvent acrale. L’histologie du MF est marquée par un infiltrat lymphocytaire dermique superficiel en bande épidermotrope riche en cellules à noyau cérébriforme. Figure 1. Mycosis fongoïde en plaques non infiltrées. Dans l’épiderme, les cellules tumorales sont groupées en amas, appelés microabcès de Pautrier ou thèques. L’infiltrat tumoral est exclusivement intra-épidermique dans la maladie de Woringer-Kolopp. L’immunomarquage montre que cet infiltrat T est majoritairement CD3+, CD4+, CD45RO+ dans l’épiderme (population tumorale), CD8+ dans le derme (population réactionnelle), présentant des pertes de marqueurs pan-T (CD5, CD7 par exemple). La recherche d’un clone T majoritaire par PCR est positive dans la peau, ce qui peut aider au diagnostic différentiel dans des situations anatomocliniques difficiles. Il peut exister un clone T circulant, surtout dans les stades avancés, ce qui constitue un élément pronostique péjoratif. Le marqueur CD158k ou KIR3DL2 est un nouveau marqueur des cellules de Sézary(4), permettant d’identifier les cellules tumorales dans la peau ou dans le sang, mais ce marqueur n’est pas encore utilisé en routine. La présence d’un clone T circulant est un élément pronostique péjoratif. Syndrome de Sézary Le syndrome de Sézary (SS), de nettement moins bon pronostic que le MF, est défini cliniquement par une érythrodermie souvent oedémateuse, volontiers associée à une kératodermie palmoplantaire et une alopécie, et biologiquement par la circulation leucémique de plus de 1 000 cellules de Sézary/mm3 ou l’association d’un clone T positif et d’une hyperlymphocytose. L’histologie est la même que celle du MF, même si l’infiltrat est souvent moins dense et un peu moins épidermotrope. La présentation clinique du mycosis fongoïde peut être polymorphe. Le diagnostic est anatomo-clinique. Staging du lymphome Le pronostic du MF et du SS dépend du stade TNMB (tableaux 2 et 3). L’extension (< ou > 10 % de la surface corporelle) et l’aspect des lésions cutanées (plaques, tumeurs ou érythrodermie) définit le T. La présentation d’emblée tumorale d’un MF est maintenant plutôt considérée comme un LTC non MF qui sera diagnostiqué selon l’aspect histologique dans la classification OMS-EORTC. L’apparition de lésions tumorales (Illustration) dans l’évolution d’un MF doit faire pratiquer de nouvelles histologies cutanées pour rechercher une transformation histologique en lymphome à grandes cellules (plus de 25 % de grandes cellules dans l’infiltrat). L’existence de ganglions palpables de plus de 1,5 cm de diamètre impose la réalisation d’une biopsie ganglionnaire pour différencier un ganglion dermopathique (N1) d’un ganglion infiltré tout ou partie par le processus tumoral (N2, N3). En cas de suspicion de métastase viscérale, une histologie est souhaitable pour prouver la nature lymphomateuse du processus d’envahissement (M1). Ce sont surtout les stades T3 (tumeurs) et T4 (érythrodermie) qui peuvent se compliquer de localisations extracutanées. Un stade B a été ajouté à la classification TNM princeps, prenant en compte la notion de circulation leucémique de cellules de Sézary et le clone sanguin éventuel. La présence de quelques cellules de Sézary (B0-B1) n’est pas spécifique du SS. Elle peut se voir dans un MF tumoral ou érythrodermique, voire dans certaines dermatoses très inflammatoires, car ces cellules ne sont définies que par un aspect cytologique que peuvent prendre certains lymphocytes activés. Par contre, dans ce cas, la présence d’un clone T circulant, s’il est associé et identique au clone cutané, sera un argument important pour le diagnostic de lymphome (MF, essentiellement stade T3-T4, ou SS). La présence de plus de 1 000 cellules de Sézary circulantes (B2), ou un clone T avec hyperlymphocytose ou un rapport CD4/CD8 > 10 avec hyperlymphocytose et perte de marqueurs pan-T définissent le SS. La présence de quelques cellules de Sézary (B0-B1) n’est pas spécifique du syndrome de Sézary. Quel bilan ? Le bilan à réaliser chez un patient suspect de MF ou de SS, destiné à établir le staging qui ensuite guidera la décision thérapeutique et établira le pronostic, est le suivant (2) : – biopsie cutanée pour examen anatomo-pathologique, immunomarquage avec marqueurs pan-T, parfois complétés par CD30, recherche de clone T cutané (surtout si doute histologique) ; – stade T1 ou T2 : radiographie pulmonaire ; on peut également discuter une échographie abdominale ; – autres stades T : scanner thoraco- abdomino-pelvien ; – existence d’un ganglion palpable de plus de 1,5 cm : biopsie ganglionnaire avec immunomarquages ; – examens sanguins : NFS, recherche de cellules de Sézary (sur frottis sanguin ou par cytométrie de flux – CD4+/CD7- ou CD4+/CD26-), clone T sanguin, bilan hépatique, LDH ; – l’examen complet de l’état général du patient et de ses comorbidités sera à prendre en compte dans la décision thérapeutique (par exemple, interféron contre-indiqué si insuffisance cardiaque sévère). Le pronostic du mycosis fongoïde dépend du stade TNMB déterminé par le bilan d’extension de la maladie. Pronostic (5) Les études publiées sont nombreuses et les facteurs pronostiques bien identifiés. Le stade T de la classification TNM est le meilleur facteur pronostique. Certains critères histologiques ont une valeur péjorative, comme la présence d’une mucinose folliculaire, indépendamment du stade clinique. • Stade T1 : la survie est identique à la population générale. Le risque de progression vers un stade plus avancé est estimé à 4-10 % à 5 ans, 10-13 % à 10 ans, 16 % à 20 ans. Le problème majeur est celui des récidives après traitement (75 %). • Stade T2 : la survie spécifique (décès imputés à la maladie) est de 96 % à 5 ans et de 83 % à 10 ans. Le risque relatif de décès est de 2,3 et le risque de progression vers un stade plus avancé est de 22 % à 5 ans, 32-39 % à 10 ans et 40 % à 20 ans. L’épaisseur histologique des lésions est un facteur pronostique important. • Stade T3 : le pronostic est beaucoup plus sombre et l’atteinte extracutanée fréquente. La survie spécifique est de 80 % à 5 ans et de 40 % à 10 ans s’il n’y a pas d’extension extracutanée. La survie observée (toutes causes de décès confondues) n’est que de 40 % à 5 ans. Le risque relatif de décès est de 4,7 ans par rapport à la population contrôle. • Stade T4 : les études réunissant les MF érythrodermiques et les SS ont montré un taux de survie observé de 41 % à 5 ans, 24 % à 10 ans et 7 % à 20 ans. La médiane de survie observée est de 3,6 ans. Les trois facteurs pronostiques majeurs sont un âge > 65 ans, un taux de cellules de Sézary circulantes > 5 % du nombre total de lymphocytes et/ou un clone T circulant et l’existence d’une atteinte ganglionnaire ou viscérale. La fréquence de survenue d’une extension ganglionnaire ou viscérale augmente avec la sévérité de l’atteinte cutanée (rare si T2, nettement plus importante si T3 ou T4). Cette extension extracutanée assombrit très nettement le pronostic : 30 % de survie spécifique à 5 ans, médiane de survie 1,5 an. Autres lymphomes T cutanés (1,3) Beaucoup plus rares, ils sont définis par un leur aspect anatomopathologique. La papulose lymphomatoïde est la plus bénigne de ces affections, avec un aspect clinique souvent caractéristique fait de lésions papulo-nécrotiques évoluant par poussées spontanément régressives (figure 2). La transformation en lymphome agressif est exceptionnelle. Les autres LTC non MF et non SS réalisent des nodules et/ou des tumeurs, uniques ou multiples, parfois ulcérés, ou bien un aspect de panniculite. Ce sont les caractéristiques anatomo-pathologiques qui permettront le diagnostic nosologique selon la classification OMS-EORTC. Figure 2. Papulose lymphomatoïde. Un bilan d’extension est nécessaire pour prouver le caractère primitivement cutané de la lymphoprolifération, préciser le stade (tableau 4) et donc guider le choix thérapeutique. La présentation clinique des lymphomes T cutanés non MF/SS est polymorphe. L’analyse histologique et immuno-histochimique est indispensable à l’établissement du diagnostic et du pronostic. Le bilan à réaliser devant un LTC autre que MF/SS est le suivant (3) : – bilan sanguin : NFS-plaquettes, LDH, phénotypage lymphocytaire en cas d’hyperlymphocytose, recherche de clone T dans le sang ; – imagerie : scanner thoracoabdomino- pelvien, scanner du cou si besoin, éventuellement complétés dans certains cas par un FDG-PET-scan ; – biopsie ostéo-médullaire : indiquée surtout pour les LTC agressifs, discutée pour les LTC indolents ; – cytoponction ganglionnaire, voire biopsie ganglionnaire au moindre doute si adénopathie clinique > 1 cm et/ou fixant au PETscan. Pronostic des LTC non MF et non SS (5) Les études pronostiques sont peu nombreuses. Sont de bon pronostic : • Papulose lymphomatoïde : survie spécifique 100 % à 5 ans. • Autres lymphomes T à grandes cellules CD30+ (hors transformation histologique de MF) : survie spécifique 96 % à 5 ans. Cependant, l’existence de tumeurs multifocales lors du diagnostic serait un facteur péjoratif. • Lymphome T sous-cutané à type de panniculite : il se présente comme une plaque infiltrée ou des nodules des membres inférieurs, simulant une panniculite. Histologiquement, l’infiltrat est hypodermique, de phénotype αβ CD8+. La survie à 5 ans est de 80 %. • Lymphome T pléomorphe à petites et moyennes cellules CD4+ : il se présente comme des plaques, nodules ou tumeurs ; histologiquement, l’infiltrat est profond et non épidermotrope. La recherche d’un clone T est capitale pour le diagnostic différentiel avec un pseudolymphome. La survie à 5 ans est de 60-80 %. Sont de mauvais pronostic : • Lymphome T à grandes cellules CD30- : il se présente sous forme de nodules et tumeurs de croissance rapide avec dissémination extracutanée rapide. La survie spécifique à 5 ans est inférieure à 20 %. • Lymphome T épidermotrope CD8+ : cliniquement, il s’agit de papules, nodules ou tumeurs ulcéro-nécrotiques d’extension rapide. La médiane de survie est de 32 mois. • Lymphome T γδ: il a la même présentation clinique que le LTC épidermotrope CD8+ avec possibilité de lésions plus profondes de type panniculite et souvent plus disséminées. Le syndrome d’hémophagocytose est une complication fréquente. Histologiquement, l’infiltrat est de topographie variable, plus ou moins profond, et est constitué de lymphocytes T cytotoxiques γδ. La médiane de survie est de 15 mois. Traitement des LTC Le traitement du MF, du SS, de la papulose lymphomatoïde et du lymphome à grandes cellules CD30+ a fait l’objet d’un algorithme thérapeutique proposé par un groupe d’experts (6). Depuis, de nouveaux traitements arrivent sur le marché, comme l’alemtuzumab (Campath®), la forodésine et les inhibiteurs d’histone désacétylases, pour l’instant réservés aux situations d’échec thérapeutique (tableau 5). Les objectifs du traitement aux stades précoces sont l’obtention d’une rémission complète durable et la préservation d’une qualité de vie optimale. Aux stades avancés, il n’existe aucun traitement curateur et l’objectif sera alors de prolonger la survie et d’obtenir une réponse partielle ou plus rarement complète, parfois un simple contrôle palliatif de la maladie, par l’utilisation alternée des différents moyens thérapeutiques disponibles, en respectant autant que possible la qualité de vie par un contrôle des effets indésirables des traitements. L’alemtuzumab est un anticorps monoclonal humanisé dirigé contre CD52, un antigène exprimé par les lymphocytes normaux et tumoraux. Il s’administre par voie sous-cutanée et a montré des taux de réponse supérieurs à 50 %, dont plus de 30 % de réponses complètes dans des MF graves ou des SS. La complication majeure est la survenue possible d’infections opportunistes graves (cytomégalovirus, toxoplasmose, etc.). Les inhibiteurs des histone désacétylases (iHDAC) représentent une nouvelle classe médicamenteuse actuellement en plein essor pour les LTC réfractaires. Ils agissent en modifiant l’état d’acétylation des histones, protéines de la chromatine autour desquelles le double brin d’ADN s’enroule. Cette modification du statut d’acétylation joue un rôle important dans la régulation de la transcription et dans la réparation de l’ADN. Les iHDAC peuvent induire une réversion du phénotype tumoral et une apoptose des cellules tumorales (mais pas des cellules normales). Le chef de file est le suberoylanilide hydroxamic acid (SAHA) ou vorinostat (Zolinza®), qui s’administre per os. Le taux de réponse est de 24 % dans une étude ouverte de phase 2 avec une durée de réponse de 15 semaines. Le prurit semble bien amélioré par le traitement. Les modalités thérapeutiques du MF sont bien codifiées et dépendent du stade TNM. De nouvelles classes thérapeutiques sont en développement pour les formes avancées de la maladie. Conclusion Le pronostic des lymphomes T cutanés dépend du type histologique et de l’extension de la maladie. De nombreux traitements sont aujourd’hui disponibles mais aucun n’est actuellement définitivement curateur. De nouvelles molécules sont en développement pour les formes réfractaires les plus graves et devraient permettre d’élargir l’arsenal thérapeutique. L’EORTC Cutaneous Lymphoma Task Force, groupe européen d’étude des lymphomes cutanés, développe une plateforme thérapeutique comportant trois protocoles comparatifs successifs. L’inclusion des malades ayant un MF/SS au stade avancé dans de tels protocoles est le seul moyen de faire progresser la prise en charge thérapeutique de ces lymphomes.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :