Publié le 06 jan 2009Lecture 3 min
Le syndrome de Woringer-Kolopp
S. DIKHAYE, B. HASSAM, Service de dermatologie et de vénéréologie, CHU Ibn-Sina, Rabat, Maroc
Le syndrome de Woringer-Kolopp ou lymphome pagétoïde a été décrit en 1939 par Frédéric Woringer et Pierre Kolopp, Otto Braun-Falco et ses collaborateurs ont décrit la réticulose pagétoïde (ancienne dénomination)(1,2,3). Il s’agit d’une variante du mycosis fongoïde de plutôt bon pronostic.
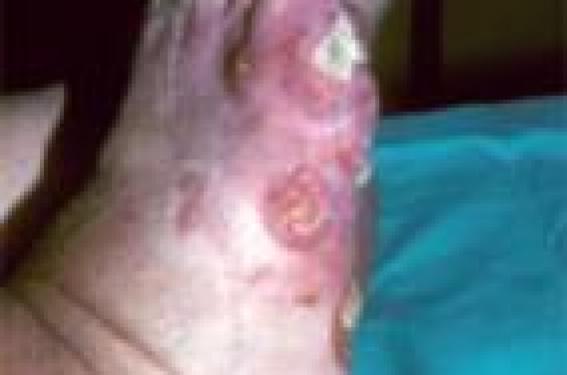
Dans la nouvelle classification des lymphomes cutanés, la réticulose pagétoïde désigne uniquement le syndrome de Woringer-Kolopp qui est considéré comme étant une variante du mycosis fongoïde(4) (caractérisée par son aspect localisé et par un épidermotropisme massif) ; la forme disséminée type Ketron-Goodman en est exclue. Dans sa forme caractéristique, il se manifeste cliniquement par une lésion unique en plaque érythémato- squameuse psoriasiforme, bien limitée, hyperkératosique voire verruqueuse, la bordure est polycyclique avec tendance à la guérison centrale. Elle siège le plus souvent au niveau de l’extrémité d’un membre. Son évolution est lente et il n’a jamais été rapporté de localisation extracutanée (figure 1). Figure 1. Lésions nodulaires et ulcéro-bourgeonnantes du pied dans le cadre d’un syndrome de Woringer-Kolopp. La forme caractéristique se manifeste par une lésion unique en plaque érythémato-squameuse psoriasiforme. Histologie Histologiquement, il se caractérise par une infiltration par des cellules atypiques pagétoïdes. L’épidermotropisme de l’infiltration est massif, avec une acanthose et une désagrégation des couches basales de l’épiderme. Les cellules de l’infiltrat sont de taille moyenne à grande (15 à 20 mm) ; dans l’épiderme, elles sont isolées ou regroupées en nids (en thèques) ou en amas, d’où le terme de pagétoïde (figure 2). Leur noyau est quelquefois hyperchromatique et convoluté, et leur cytoplasme est abondant et vacuolisé. L’infiltrat dermique est plus polymorphe que celui observé dans le mycosis fongoïde. Il occupe toute la hauteur de l’épiderme tout au long de la lésion(5) (figure 2). Les cellules tumorales sont de phénotype le plus souvent CD4+, CD8-, CD3+, CD2+ mais parfois, elles sont CD4-, CD8+ voire CD4-, CD8-, l’antigène CD30 est souvent exprimé(3). De manière très rare, une perte de l’expression de l’antigène CD7 a été rapportée. Il existe habituellement un réarrangement clonal des gènes codant le récepteur T lymphocytaire. Traitement Le traitement est celui d’un mycosis fongoïde dans une forme localisée : – chimiothérapie locale : carmustine (BCNU) ou méchloréthamine (caryolysine) ; – radiothérapie externe localisée aux alentours de 20 grays(6) ; – PUVAthérapie ; – de manière beaucoup plus exceptionnelle, l’excision chirurgicale simple(7), avec une marge de 0,5 cm peut être une alternative ; – enfin, la photothérapie dynamique topique (PDT)(8) demeure du domaine de la recherche clinique. Le pronostic est excellent. Il n’a jamais été rapporté de localisations extracutanées. Cependant, des récurrences locales après traitement peuvent survenir. Figure 2. Épidermotropisme massif et infiltration par des cellules atypiques pagétoïdes. Avec l’aimable collaboration de D. BLANC
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :