Publié le 18 fév 2009Lecture 10 min
Diagnostic de sclérodermie devant une polyarthralgie à doigts boudinés
C. TOLEDANO, service de médecine interne, hôpital Saint-Antoine, Paris
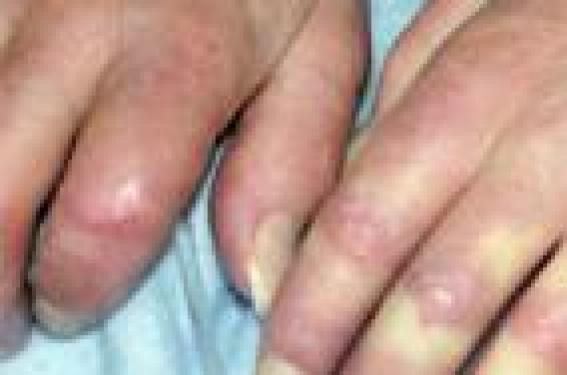
La sclérodermie systémique est considérée actuellement comme la plus grave des connectivites car elle a le plus mauvais pronostic. Le diagnostic des formes évoluées avec sclérose cutanée proximale est aisé. La reconnaissance des formes débutantes à l’aide de quelques signes cliniques (sclérodactylie, arthralgies, syndrome de Raynaud suspect, télangiectasies, calcifications sous-cutanées et reflux gastro-oesophagien) aidé d’examens complémentaires simples (recherche d’auto-anticorps, capillaroscopie, radiographie pulmonaire et radiographies des mains), pourrait permettre de prendre en charge la maladie à un stade précoce et d’en améliorer à terme le pronostic.
La sclérodermie systémique (ScS) est une maladie de physiopathologie complexe se traduisant par une microangiopathie sclérosante avec accumulation de fibres de collagène dans la peau et les organes profonds. Sa prévalence est estimée à 200 à 300 cas par million d’habitants, elle touche la femme 8 fois sur 10. Son pronostic est fortement associé à l’atteinte des organes profonds elle-même corrélée à l’évolutivité des lésions cutanées avec une survie à 9 ans évaluée entre 38 % et 72 % selon qu’il existe ou pas une atteinte sévère d’organe (1). La ScS est donc considérée comme la maladie de système ayant le plus mauvais pronostic car elle présente le taux de survie le plus faible. En comparaison les taux de survie du lupus systémique ou de la polyarthrite rhumatoïde sont actuellement supérieurs à 90 % à 5 ans. L’atteinte sévère d’organe (poumon, rein, coeur) survient précocement au cours de la maladie chez les patients développant la forme cutanée diffuse : 70 % des atteintes rénales ou cutanées sévères et 50 % des atteintes cardiaques, interstitielles pulmonaires et digestives sont diagnostiquées au cours des 3 premières années d’évolution1. Ce pronostic incite donc à un diagnostic précoce de la maladie et de l’atteinte des organes profonds ainsi qu’à une surveillance rapprochée pour espérer à terme ralentir l’évolution de la maladie. Classification Les critères de l’ACR, critères diagnostiques considérés comme des critères de classification, individualisent 2 formes principales de ScS (2) : soit le patient présente une infiltration cutanée proximale (critère majeur), soit il doit réunir 2 critères mineurs (sclérodactylie/ulcération pulpaire/cicatrices des pulpes digitales/fibrose pulmonaire des 2 bases). Le problème principal de ces critères est leur manque de sensibilité, et surtout leurs mauvaises performances diagnostiques dans les formes débutantes et cutanées limitées. Une classification plus récente a tenté d’identifier les formes précoces et les formes sans atteinte cutanée proximale, elle prend en considération le phénomène de Raynaud, la capillaroscopie et les anticorps spécifiques (3) (tableau ci-dessous). ScS limitée • Phénomène de Raynaud objectif + : - soit anomalie capillaroscopique - soit anticorps spécifiques ScS cutanée limitée (ex. syndrome CREST) • Critères précédents + infiltration cutanée distale (en aval des coudes et genoux) Clinique à la phase précoce La présentation clinique majoritaire en phase de début associe en général, à des degrés variables, un syndrome de Raynaud qui peut précéder les autres symptômes de la maladie de plusieurs années, des doigts boudinés (sclérodactylie) avec arthralgies, une atteinte de la partie basse de l’oesophage se manifestant par un reflux gastro- oesophagien et parfois des signes de fibrose interstitielle des bases pulmonaires. Les atteintes cutanées Figure 1. Patiente atteinte de sclérodermie systémique : syndrome de Raynaud, sclérodactylie, doigts bloqués en flexion. Sclérodactylie (figure 1) Atteinte cutanée inaugurale la plus fréquente, elle évolue progressivement selon 3 phases : • oedémateuse avec épaississement des extrémités, les doigts deviennent « boudinés » ; • induration, les doigts s’effilent ; • atrophie : la peau adhère au squelette, il apparaît des ulcérations pulpaires douloureuses invalidantes et les ongles deviennent dystrophiques. À un stade ultérieur, les doigts puis la main se figent en flexion, formant les « mains en griffes ». L’atteinte cutanée évolue ensuite vers une ScS beaucoup plus facile à identifier avec acrosclérose puis sclérose diffuse. Télangiectasies Elles sont très fréquentes au cours de la ScS, correspondent à des dilatations des veinules et des capillaires. Elles sont présentes surtout sur le visage, les lèvres, la langue et les paumes. Le phénomène de Raynaud L’ischémie paroxystique des extrémités déclenchée principalement par le froid ou plus rarement par les émotions est présente chez environ 15 % des femmes et 10 % des hommes. La forme typique évolue en trois phases : • blanche (ischémie artérielle) : presque toujours présente, de survenue brutale, les doigts deviennent blancs, froids, insensibles, avec une limite supérieure nette au niveau des phalanges ou du dos de la main ; • bleue (stase) asphyxique avec cyanose des doigts ; • rouge (vasodilatation réactionnelle) ; il existe alors une hyperhémie, douloureuse jusqu’à la recoloration normale. Le syndrome de Raynaud est le plus souvent bilatéral, symétrique, et peut toucher les pieds, le nez, les oreilles, ou la langue. Plus de 90 % des phénomènes de Raynaud sont primitifs. L’interrogatoire et l’examen clinique recherchent des causes favorisantes comme les activités professionnelles à risque (syndrome du marteau hypothénar, la maladie des vibrations), les médicaments favorisants (dérivés d’ergot de seigle, bêtabloquant, etc.), un syndrome du canal carpien, ou un syndrome de la traversée thoracobrachiale. Ces deux dernières étiologies ne doivent cependant pas dispenser d’un bilan à la recherche de ScS. On éliminera également les diagnostics différentiels représentés par les autres acrosyndromes vasculaires (acrocyanose, acrorighose, érythomélalgie). Devant un syndrome de Raynaud, on cherchera les éléments cliniques permettant de suspecter un syndrome de Raynaud secondaire, en particulier : • un phénomène de Raynaud parfois très sévère associé à des troubles trophiques digitaux (figures 2 et 3). La présence de cicatrices pulpaires déprimées (à rechercher à l’examen minutieux des pulpes), la présence d’ulcération pulpaire ; • absence de rémission estivale ; • début après 40 ans ; • absence d’antécédent familial de maladie de Raynaud ; • atteinte de tous les doigts (y compris le pouce) ; • présence d’autres signes cliniques de ScS ; • manoeuvre d’Allen positive. Figure 2. Sclérodermie systémique : ulcération pulpaire (5e doigt) ; cicatrices pulpaires (2e, 3e et 4e doigts). Figure 3. Sclérodermie systémique : nécrose pulpaire. Capillaroscopie péri-unguéale Figure 4. Capillaroscopie : désorganisation du lit capillaire (capillaires élargis). La visualisation in vivo des capillaires du derme dont les anses se déroulent parallèlement au plan cutané au pourtour de l’ongle permet le dépistage de microangiopathie organique. • Un examen normal montre : - des capillaires fins homogènes avec une anse de réflexion formant une boucle en épingle à cheveux : on en compte environ 8 à 10 au mm, de 6 à 15 μm de diamètre, parallèles, régulièrement espacées et disposées sur 3 rangées. • Un examen de microangiopathie peut montrer (figure 4) : - des microhémorragies témoignant d’une souffrance capillaire ; - des dystrophies (une proportion supérieure à 15 % des anses et un aspect richement ramifié sont les témoins d’une néogenèse capillaire microangiopathie) ; - des mégacapillaires de diamètre supérieur à 50 μm avec une diminution hétérogène de la densité capillaire (plages désertes) et une désorganisation de l’arrangement des boucles sont les signes d’une micro-angiopathie organique spécifique, pathognomonique des connectivites. C’est un élément sensible et précoce, le meilleur dépistage des phénomènes de Raynaud à risque d’évolution sclérodermique. Atteinte articulaire dans la ScS débutante L’atteinte articulaire de la ScS peut être inaugurale dans 12 à 65 % des cas et apparaît au cours de l’évolution chez plus de 50 % des patients (4). Les symptômes ostéo-articulaires d’appel sont surtout des arthralgies et s’accompagnent le plus souvent d’une raideur des doigts, des mains ou des poignets, parfois associée à des synovites. On peut également rencontrer d’authentiques oligo- ou polyarthrites à prédominance distale avec signes inflammatoires locaux et raideur matinale, pouvant atteindre avec prédilection les petites articulations des mains, des poignets et des chevilles. Cette véritable oligo- ou polyarthrite est habituellement non érosive sauf lors des syndromes de chevauchement entre la ScS et la polyarthrite rhumatoïde, et peut poser un problème diagnostique avec un rhumatisme inflammatoire débutant en l’absence d’autres signes de ScS. Atteinte osseuse Figure 5. Sclérodermie systémique avec acro-ostéolyse. L’atteinte osseuse caractéristique de la ScS que l’on peut rencontrer dès le stade précoce de la maladie correspond à une résorption osseuse particulière avec acroostéolyse (figure 5), notamment au niveau des phalanges digitales terminales. On l’observe chez 29 à 80 % des patients, son mécanisme est indéterminé, mais il pourrait être en rapport avec les complications vasculaires propres à la maladie. Elle débute sur la face palmaire avec une possible destruction complète de la phalange distale ; elle est plus rare au niveau des phalanges intermédiaires des doigts, des orteils, du carpe, des avant-bras, des côtes ou de la mandibule, etc. Ces résorptions osseuses s’accompagnent souvent de calcifications sous-cutanées. Calcinose (figures 6 et 7) Elle est formée de dépôts de cristaux d’hydroxyapatite dans les tissus mous sous-cutanés et peut survenir tôt dans la maladie. Elle est présente chez 20 à 30 % des patients et prédomine aux mains ainsi qu’au niveau des zones de microtraumatismes (avant-bras, coudes). Son importance augmente avec la durée et la sévérité de la maladie, sa physiopathologie est méconnue. Ces nodosités de dimensions et formes variables (de quelques micromètres à plusieurs centimètres) sont le plus fréquemment indolores et de consistance ferme, adhérentes au plan profond, mais des réactions inflammatoires locales avec possible ulcération et/ou surinfection peuvent survenir. Figure 7. Sclérodermie systémique avec calcifications souscutanées. Figure 6. Sclérodermie systémique avec calcification. Autres signes cliniques lors de la phase de début • On recherchera cliniquement une atteinte des bases pulmonaires (fibrose interstitielle débutante avec présence de râles crépitants secs) ou plus fréquemment une atteinte infraclinique mise en évidence sur la radiologie, voire le scanner (syndrome interstitiel). • L’interrogatoire recherchera un reflux gastro-oesophagien. Les examens biologiques La recherche d’auto-anticorps peut apporter une aide au diagnostic de ScS au stade initial et à la classification. • Les anticorps antinucléaires sur cellules HEp2 sont présents chez 90 % des patients. • Les auto-anticorps spécifiques : - anti-centromères : existent chez 30 % des patients mais chez 65 % de ceux présentant une forme cutanée limitée, et donc associés à la survenue d’une HTAP ; - anti-topoïsomérase I (= anti- Scl70) : présents chez 20 % des patients mais dans 40 % des formes diffuses, et donc associés à la survenue d’une atteinte interstitielle pulmonaire grave et à la survie ; - anti-ARN polymérase III sont observés chez 4 à 25 % des patients et seraient associés à la forme diffuse et à la crise rénale ; - anti-Pm/Scl : présents chez 3 % des patients et associés à la survenue d’une myopathie inflammatoire (scléromyosite). Les syndromes de chevauchement On décrit de véritables syndromes de chevauchement (connectivite mixte) dans lesquels les patients présentent des éléments propres de 2 ou plusieurs connectivites réalisant une véritable entité clinique. La connectivite mixte : syndrome de Sharp Avec doigts boudinés douloureux (sclérodactylie), syndrome de Raynaud avec signes de microangiopathie organique à la capillaroscopie, télangiectasies, calcinose sous-cutanée, polyarthralgies, polyarthrite, atteinte digestive fréquente (75 %), atteinte pulmonaire et myopathie inflammatoire. La présence d’auto-anticorps anti-U1 RNP est un critère obligatoire du diagnostic. Ce syndrome peut évoluer vers une authentique ScS. Scléromyosites Syndrome de chevauchement le plus fréquent : 29 % des patients avec myopathies inflammatoires présentent des signes de ScS (phénomène de Raynaud, sclérodactylie, faciès sclérodermique, atteinte oesophagienne, pneumopathie infiltrante diffuse) et des signes de dermatomyosite (associés à l’atteinte musculaire modérée, peu déficitaire, avec myalgies et élévation des CPK ainsi que la présence possible d’auto-anticorps anti- PM Scl. En pratique Devant une polyarthralgie avec doigts boudinés, il est donc important d’identifier les « états présclérodermiques ». Après un interrogatoire (recherche de reflux gastro-oesophagien, syndrome de Raynaud) et un examen clinique minutieux (recherche de cicatrices pulpaires), quelques examens simples permettent d’envisager le diagnostic de ScS : la capillaroscopie, la radiographie des mains, des poumons et les auto-anticorps. Cette reconnaissance des formes précoces devrait permettre à terme d’améliorer le pronostic de cette connectivite.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



