Maladie de système, Médecine interne
Publié le 22 oct 2007Lecture 5 min
Dermatoses bulleuses auto-immunes
D. Fahri
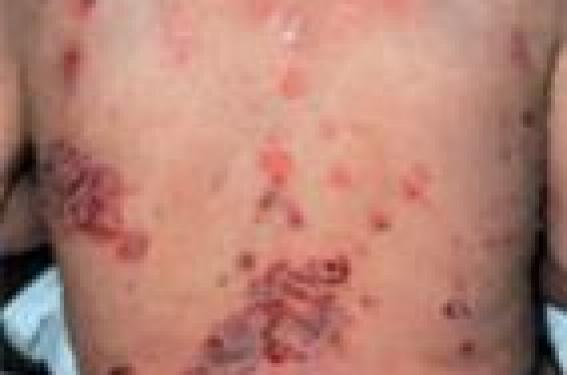
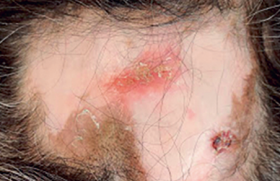
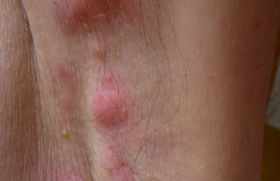
Diagnostic des dermatoses bulleuses auto-immunes : l’immunofluorescence directe (1) Dans le cadre d’une suspicion de dermatose bulleuse auto-immune (DBAI), quatre critères cliniques plaident fortement en faveur d’une pemphigoïde bulleuse (PB) : – absence d’atrophie cutanée ; – absence d’atteinte muqueuse ; absence d’atteinte cervico-céphalique ; – âge > 70 ans. L’immunofluorescence indirecte sur peau humaine clivée permet de distinguer deux groupes de DBAI avec clivage sous-épidermique, selon le niveau du dépôt d’Ig : – fixation sur le toit de la bulle : PB, pemphigoïde gestationnelle, pemphigoïde cicatricielle (PC) à Ac anti-BP180 (collagène XVII), dermatose à IgA linéaires et pemphigoïde à Ac anti-plectine ; – fixation sur le plancher de la bulle : PC à Ac anti-épiligrine ou à Ac anti-laminine 5, pemphigoïde à Ac anti-p200, épidermolyse bulleuse acquise (EBA) et lupus bulleux (anticorps anti-collagène VII). L’examen de référence, en pratique réservé aux cas de diagnostic difficile, est l’immunomicroscopie électronique. L’IFD est néanmoins un examen précieux permettant de faire le diagnostic dans la majorité des cas. La forme du dépôt jonctionnel d’IgG permet de classer les DBAI en deux groupes : – dépôts formant des créneaux en « N » dans la PB, la PC et la dermatose à IgA linéaires ; – dépôts formant des créneaux en « U » dans l’EBA et le lupus bulleux (2). Par ailleurs, un dépôt linéaire évoque une PB, une PC, une EBA ou un lupus bulleux, tandis qu’un dépôt granuleux évoque une dermatite herpétiforme. Pour une analyse de meilleure qualité, la biopsie cutanée pour IFD doit être transportée au laboratoire dans les 24 heures dans du sérum NaCl 9‰, plutôt que dans du liquide de Michel ou dans l’azote liquide. Stratégies thérapeutiques dans trois dermatoses bulleuses auto-immunes (3) Les choix thérapeutiques dans les dermatoses bulleuses autoimmunes (DBAI) dépendent du terrain (âge physiologique, comorbidités) et de la gravité (étendue) de la DBAI. La pemphigoïde bulleuse (PB) survient surtout après 70 ans (moyenne : 80 ans) et sa mortalité à 1 an est de 25 à 40 %. Le pemphigus et la pemphigoïde cicatricielle (PC) surviennent surtout entre 50 et 60 ans. La mortalité du pemphigus est d’environ 5 % sous traitement. La PC présente avant tout un risque fonctionnel (cécité). Le pronostic varie donc considérablement selon les DBAI, allant des formes localisées aux formes potentiellement létales, en passant par les formes avec risque fonctionnel. Pemphigoïde bulleuse Le taux de contrôle de la PB à J21 sous clobétasol propionate et prednisone orale est, respectivement, de 100 % et 85 % dans les formes limitées (≤ 10 bulles) et de 99 % et 91 % dans les formes étendues (> 10 bulles). Le clobétasol présente cependant des inconvénients : observance parfois difficile à obtenir, possibles effets indésirables locaux et généraux. En pratique pratique, la PB est traitée en première ligne par le clobétasol propionate (Dermoval®) crème (10 à 40 g/j en traitement d’attaque, selon l’étendue des lésions). En seconde intention, la prednisone orale sera prescrite en privilégiant des doses de 0,5mg/kg/j (à celles d’1mg/kg/j). En troisième ligne, les PB réfractaires (très rares) ou corticodépendantes (moins rares) peuvent être traitées par adjonction d’immunosuppresseur. Cependant, le bénéfice des immunosuppresseurs en termes d’épargne des corticoïdes n’a jamais été démontré dans des essais contrôlés randomisés. Les pemphigus limités peuvent être traités en première ligne selon le protocole Lever (prednisone 40 mg/j + azathioprine 100 mg/j, en traitement d’attaque). Le délai pour obtenir l’épithélialisation est habituellement long (3 à 6mois). Les formes étendues sont traitées par prednisone orale (1 à 1,5 mg/kg/j). Le délai pour obtenir l’épithélialisation est habituellement plus court (4 à 6 semaines). Lorsque le contrôle du pemphigus est obtenu, le schéma de décroissance de la prednisone suivant peut être proposé : - 15 %/15 jours entre 1,5 et 1 mg/kg/j ; puis -10 %/15 jours entre 1 et 0,2 mg/kg/j, puis - 5 %/15 jours entre 0,2 et 0,1mg/kg/j. Les formes réfractaires de pemphigus sont traitées par adjonction à la prednisone d’un immunosuppresseur, des immunoglobulines polyvalentes intraveineuses ou du rituximab (Acmonoclonal anti-CD20). Le taux de rémission complète des pemphigus sévère sous rituximab a été estimé à environ 80 %. Le taux de rechute était de 45 % avec un délai médian de 18 mois. Pemphigus Pemphigoïde cicatricielle La PC et l’épidermolyse bulleuse acquise répondent habituellement mal aux corticoïdes oraux. Les formes limitées de PC sont traitées par disulone (75 à 150 mg/j), dermocorticoïdes forts ou tacrolimustopique. Les formes sévères (avec atteintes oculaires ou aérodigestives supérieures menaçantes, notamment) seront traitées par corticoïdes oraux (0,5 mg/kg/j), disulone et cyclophosphamide (100 mg/j per os ou 1 g/mois, IV). Les données disponibles sur l’efficacité du mycophénolate mofétil, des Ig polyvalentes IV et des biothérapies (rituximab, anti-TNF) dans la PC sont limitées.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :