Maladie de système, Médecine interne
Publié le 05 jan 2020Lecture 2 min
Le VEXAS : une nouvelle maladie auto-inflammatoire de l’adulte que doit connaître le dermatologue

Il existe de nombreuses maladies auto-inflammatoires monogéniques, avec chaque année de nouvelles mutations découvertes et des nouveaux syndromes. Ces maladies sont aujourd’hui classées en fonction des voies moléculaires identifiées et des phénotypes en particulier les interféronopathies, les maladies de l’inflammasome, les RAS-opathies ou les NF-kB-opathies… L’immense majorité de ces pathologies débutent dans l’enfance et les patients présentent souvent des signes dermatologiques qui peuvent être au premier plan.
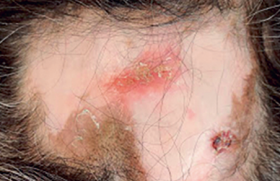
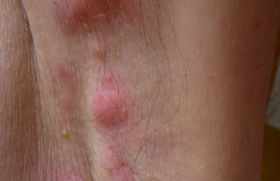
Les maladies inflammatoires survenant chez l’adulte sont beaucoup plus rares. Depuis plusieurs années, plusieurs équipes ont rapporté des maladies inflammatoires ou auto-immunes survenant chez des patients présentant un syndrome myélodysplasique ou myéloprolifératif, mais le lien direct entre les deux pathologies restait à démontrer (sauf peut-être pour le syndrome de Sweet histiocytoïde et les syndromes myélodysplasiques, ou certaines histiocytoses avec des leucémies myélomonocytaires chroniques). Un article récemment publié dans le New England Journal of Medicine a décrit un nouveau syndrome auto-inflammatoire nommé VEXAS (vacuoles, E1 en - zyme, X-linked, auto-inflammatory, somatic). En effet, les auteurs ont identifié chez 25 hom mes, des mutations somatiques du gène UBA1, un gène nécessaire à l’ubiquitination (rôle nécessaire à la dégradation protéique) localisé sur le chromosome X. Ces mutations étaient postzygotiques, touchant les populations myéloïdes avec des fréquences de variant alléliques mutés > 70 % chez les malades. Trois mutations ont été identifiées, toutes touchant la partie p.Met41, résultant à une perte d’activité cytoplasmique de la protéine. En microscopie électronique, les cellules myéloïdes avaient des vacuoles (gouttes lipidiques) signes de mort cellulaire. Cliniquement, les patients (tous des hommes) avaient un âge médian de 64 ans (extrêmes 45-80), avaient une fièvre récurrente dans 92 % des cas, une atteinte cutanée dans 88 % des cas, en particulier des syndromes de Sweet et des vascularites, des infiltrats pulmonaires dans 72 % des cas, des chondrites nasales et auriculaires dans 64 % des cas, des thromboses veineuses dans 44 % des cas. Au plan hématologique, il existait une anémie macrocytaire dans 96 % des cas et des vacuoles sur le myélogramme dans 100 % des cas (18 testés). Il existait un syndrome inflammatoire constant, avec une CRP médiane à 78 (extrêmes 18-128) et 40 % des patients sont décédés pendant le suivi de complications infectieuses ou des cytopénies. Cependant, aucun des patients n’a évolué vers une myélodysplasie avec excès de blaste ou une leucémie aigüe secondaire. De façon importante, 15 (60 %) des patients remplissaient les critères de classification de la PCA, 32 % d’un syndrome de Sweet et 24 % d’un syndrome myélodysplasique.
En utilisant un modèle animal de zebrafish, les auteurs ont confirmé que la perte d’activité de la protéine UBA1 isoforme dans le cytoplasme était responsable d’une inflammation systémique importante, confirmant le caractère pathogène de la mutation. Les patients masculins avec une PCA, âgés, un syndrome inflammatoire biologique et des anomalies hématologiques pré - sentent donc possiblement un syndrome VEXAS qu’il faut savoir rechercher.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :