Publié le 20 juin 2018Lecture 6 min
Syndrome d’activation macrophagique révélant un lupus aigu - Importance des signes dermatologiques pour un diagnostic urgent
Philippe BERBIS, Service de Dermatologie, Hôpital Nord, CHU de Marseille
–
Observation
Un homme de 39 ans, sans antécédent particulier, est admis en urgence pour une altération fébrile récente de l’état général, évoluant et s’aggravant depuis 2 mois. À l’entrée, la température, évoluant dans un premier temps par pics, puis en plateau, est à 39° C. Il présentait un état confusionnel et des polyarthralgies à la fois périphériques (chevilles, poignets) et axiales (articulations sacro-iliaques). Au plan dermatologique, il présentait des lésions inflammatoires, érythémateuses, ulcérées par en droits, recouvertes d’un enduit fibrineux et nécrotique, par endroits en cocarde (figures 1 et 2).
Figure 1. Lésion ulcérée nécrotique du pavillon de l’oreille.
Figure 2. Lésion inflammatoire en cocarde.
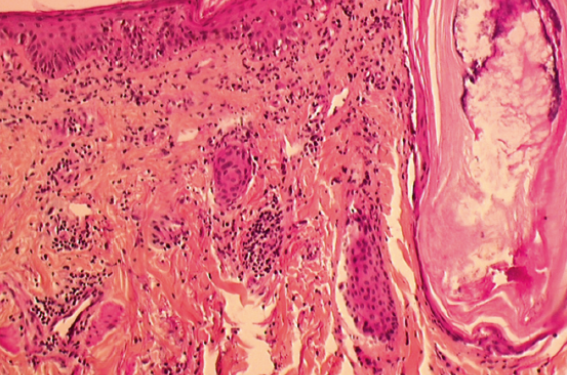
Ces lésions, très douloureuses, touchaient le visage, le cuir chevelu, les oreilles et le décolleté. Il présentait des douleurs intenses à la déglutition et un syndrome diarrhéique. Le reste de l’examen notait une hépatosplénomégalie douloureuse à la palpation. L’examen ophtalmologique, motivé par des douleurs orbitaires, montrait une vascularite rétinienne. Le bilan biologique de base montrait une leucopénie modérée, un syndrome inflammatoire majeur (PCR : 46 mg/l, VS 70/90), des LDH élevées et une légère cytolyse hépatique. La biopsie cutanée montrait un intense infiltrat inflammatoire dermique (figure 3) avec vasculite leucocytoclasique et phénomènes de nécrose épidermique.
Figure 3. Infiltrat inflammatoire avec vasculite leucocytoclasique.
L’immunofluorescence directe était positive avec aspect de bande lupique : positivité granuleuse de la basale et en périannexiel (figure 4). Le tableau clinique s’aggravait rapidement de même que les signes biologiques, avec une pancytopénie (hémoglobine à 10 g/dl, neutropénie à 0,2 g/l, thrombopénie à 73 g/l). Le myélogramme montrait une moelle infiltrée par des histiocytes non tumoraux, avec phénomènes d’hématophagocytose nets.
Figure 4. Aspect de bande lupique en mmunofluorescence directe.
Le bilan montrait par ailleurs une hyperferritinémie majeure, une forte hypertriglycéridémie, une insuffisance rénale aiguë, une aggravation de la cytolyse hépatique et une rhabdomyolyse. Le diagnostic de syndrome d’activation macrophagique (SAM) était porté et un traitement instauré en urgence : bolus de méthylprednisolone (1 g/jour pendant 3 jours) suivi d’un relais per os à forte dose (1,5 mg/kg/j), associé à des immunoglobulines intraveineuses (400 mg/kg/j pendant 5 jours). Cette prise en charge entraînait une amélioration rapide et spectaculaire du tableau, tant au plan clinique que biologique. Parallèlement, un bilan étiologique était effectué, totalement négatif au plan infectiologique en dehors d’une PCR sanguine positive pour l’EBV (Epstein Barr virus), avec une sérologie évocatrice d’une immunisation ancienne, pouvant témoigner d’une réactivation. La recherche d’une néoplasie était également négative (scanner thoraco-abdominopelvien normal en dehors d’une sacro-iléite bilatérale), endoscopie digestive normale, examen ORL normal en dehors d’ulcérations pharyngées d’allure inflammatoire. Le bilan immunitaire montrait la présence d’anticorps antinucléaires positifs (1/400e) avec anti-DNA natifs positifs, et une diminution du CH50 et du C3. La recherche d’anticorps anticytoplasmes des polynucléaires et d’anticorps antiphospholipides était négative, de même que les anticorps anti-CCP et le facteur rhumatoïde. Les marqueurs HLA associaient un phénotype HLA B27, corrélé à la sacro-iléite et B51. Au total, ce patient présentait un syndrome d’activation macrophagique (SAM) ayant révélé un lupus érythémateux aigu et une sacro-iléite HLA B27 positive. Le diagnostic de SAM était retenu sur les critères classiques définis dans le tableau ci-dessous et le lupus érythémateux aigu sur les critères de l’ACR : lésions cutanéo-muqueuses spécifiques, polyarthrite, leucopénie, anticorps antinucléaires et anti-DNA natifs positifs. Le rôle déclenchant de l’infection par EBV peut être discuté bien que l’on ne puisse écarter une réactivation au cours d’une poussée de lupus érythémateux.
Discussion
Le SAM est un syndrome hématophagocytaire lympho-histiocytaire potentiellement fatal en l’absence de prise en charge urgente. L’urgence diagnostique met en relief l’importance des signes dermatologiques(1). On distingue les SAM primaires ou familiaux et, plus fréquents, les SAM secondaires ou réactionnels. Un SAM peut ainsi révéler, ou plus fréquemment compliquer, l’évolution de pathologies néoplasiques solides, d’hémopathies (lymphomes principalement), ou de pathologies auto-immunes ou auto-inflammatoires diverses (lupus érythémateux comme dans notre observation, mais aussi sclérodermie, dermatomyosite, Gougerot-Sjögren, arthrite juvénile, maladie de Still, maladie de Kikuchi, polyarthrite rhumatoïde). Le facteur déclenchant est le plus souvent un facteur infectieux (virus dans plus de 50 % des cas, principalement virus du groupe herpes, bactéries, champignons, parasites). Un facteur déclenchant médicamenteux est la deuxième cause de SAM sur ces terrains néoplasiques ou plus souvent auto-immuns (antiinflammatoires non stéroïdiens principalement). Le diagnostic de SAM peut être difficile lorsqu’il est, comme dans notre observation, inaugural de la pathologie sous-jacente. Les signes dermatologiques ne sont pas spécifiques mais peuvent orienter le diagnostic dans un contexte biologique par ailleurs compatible : exanthème morbilliforme, lésions inflammatoires et nécrotiques cutanéo-muqueuses, lésions purpuriques, œdème facial et des extrémités, hypodermites. Au plan systémique, tous les organes peuvent être touchés selon les formes et l’intensité du tableau, principalement signes neurologiques (syndrome confusionnel, céphalées), respiratoires, hépato-splénomégalie traduisant l’infiltration par la prolifération lympho-histiocytaire. Au plan biologique, il n’existe pas de signes pathognomoniques mais un faisceau d’anomalies. Les anomalies les plus évocatrices sont une pancytopénie qui va en s’aggravant en l’absence de traitement urgent, traduisant l’hémophagocytose lympho-hitiocytaire médullaire objectivée par le myélogramme, qui, seul, permet de confirmer le diagnostic. Les autres anomalies biologiques évocatrices sont la présence d’une hypofibrinogénémie (possibilité d’une CIVD dans les formes les plus sévères), une élévation des LDH, une hyperferritinémie importante, une hypertriglycéridémie, une hyponatrémie et une cytolyse hépatique plus ou moins marquée. Le SAM est l’expression d’une prolifération incontrôlée et brutale de lymphocytes T, sécrétant de grandes quantités de cytokines, principalement IL1, IL2, IL6, IFN γ, TNFα, M-CSF. L’activation macrophagique est secondaire à cette activation lymphocytaire, et infiltre les organes hématopoïétiques, notamment la moelle osseuse, avec hémophagocytose responsable de la pancytopénie. TRAITEMENT Le traitement du SAM repose sur la mise en place d’urgence de bolus de corticoïdes en intraveineux (méthylprednisolone en bolus de 1 g/j pendant 3 jours puis relais per os à forte dose (1,5 mg /kg/j), associé à un immunosuppresseur (cyclosporine le plus fréquemment ou immunoglobulines intraveineuses comme dans notre observation). Le traitement de l’infection sous-jacente et l’arrêt d’une molécule suspecte doivent être effectués parallèlement. Le caractère original de notre observation réside dans le fait que le SAM a été inaugural chez ce patient dès l’entrée dans la maladie lupique. La survenue d’un SAM au cours du lupus érythémateux est un phénomène rare, voire très rare, puisque son incidence est estimée à 0,9 à 5 %(2). Les observations de lupus érythémateux révélés par un SAM apparaissent a fortiori exceptionnelles puisqu’une revue récente de la littérature n’en a colligé que 26 cas(2). Une étude multicentrique récente a rapporté 38 cas de SAM au cours de lupus chez l’enfant avec des caractéristiques peu diffé- rentes de celles de l’adulte(3). Le diagnostic différentiel peut être difficile avec une poussée de lupus, et il faudra alors retenir avec attention les signes cutanés et les anomalies biologiques associées à la pancytopénie : hyperferritinémie, hyponatrémie, hypertriglycéridémie, élévation du taux sérique de récepteur soluble à l’IL2. Au total, le diagnostic de SAM doit systématiquement être évoqué au cours d’une éruption fébrile associée à une cytopénie, principalement chez des patients atteints de néoplasie ou de pathologie auto-immune. La conjonction d’anomalies biologiques évocatrices doit conduire sans tarder à pratiquer un myélogramme qui seul confirmera le diagnostic et permettra la mise en route urgente d’un traitement pour éviter l’évolution fatale. Certains cas, comme dans notre observation, apparaissent particulièrement difficiles car inauguraux de la pathologie sous-jacente.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



