Maladie de système, Médecine interne
Publié le 17 mai 2016Lecture 19 min
Syndrome d’Ehlers-Danlos-Tschernogobow : nouvelles données sur le diagnostic clinique
C. HAMONET*, D. DEPARCY**, D. FRÉDY***, L. DUCRET**** - *Professeur émérite, Faculté de médecine de Créteil, consultation Ehlers-Danlos, Hôtel-Dieu, Paris ; **Service de médecine physique et de réadaptation, centre hospitalier Gustave Dron, Tourcoing ; **
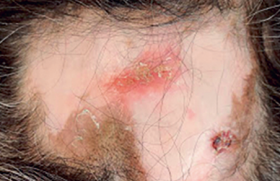
Le syndrome d’Ehlers-Danlos (SED) apparaît aujourd’hui comme une maladie familiale systémique du collagène responsable d’une diminution de la résistance et de l’élasticité du tissu conjonctif. Non ou très tardivement diagnostiquée malgré sa fréquence élevée, elle expose à des complications sévères, principalement par iatrogénie, et à de multiples situations de handicap altérant la qualité de vie.
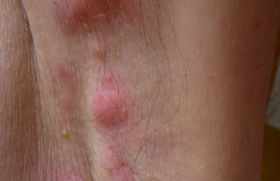
Essai de reconstitution d'une naissance contrariée Quelques rappels historiques sont nécessaires pour mieux comprendre la confusion sémiologique dans laquelle se trouve aujourd’hui le syndrome d’Ehlers-Danlos. Le 15 décembre 1900 Edvard Lauritz Ehlers fait une excellente description de ce syndrome(1) à la Société danoise de dermatologie et syphiligraphie, à Copenhague, à propos d’un cas chez un étudiant en droit. Cette description fait suite à celle du dermatologue russe Tschernogobow(2) qui, en 1892, présente deux patients à la Société de dermatologie et vénérologie de Moscou. Il décrit la fragilité de la peau, les difficultés de cicatrisation, son étirabilité, associée à une hypermobilité articulaire et des pseudo-tumeurs molluscoïdes. C’est sous le nom de ce premier descripteur oublié que les Russes désignent le SED. En 1908, Henri Alexandre Danlos(3), associé à M. Pautrier pour l’histologie, présente à la Société française de dermatologie et de syphiligraphie de Paris « un cas de Cutis laxa avec tumeurs par contusion chronique des coudes et des genoux ». Il insiste sur deux signes : « la minceur anormale » et « l’élasticité extraordinaire » de la peau qu’il compare à celle « d’une mince lame de caoutchouc ». La singularité de l’histologie de la peau de ce patient, relatée par Achille Miget(4) dans sa thèse de médecine réalisée à l’hôpital Saint-Louis en 1933, et le tableau clinique sont plutôt en faveur d’un pseudo-xanthome élastique. Par la suite, la notion d’une étirabilité importante sera considérée par bon nombre de médecins, comme un signe nécessaire au diagnostic de SED, écartant ainsi un très grand nombre de patients. Malgré cela, Miget réunira dans le même syndrome Ehlers et Danlos, trois ans avant que Frederick Parkes-Weber ne le propose en 1936. La suite de l’histoire du syndrome d’Ehlers-Danlos est marquée par l’implication de deux disciplines médicales dans sa description et ses essais d’identification. La rhumatologie avec Grahame(5) et la génétique avec Beighton. Le caractère de bénignité, l’absence de douleurs en particulier, accolée à l’hypermobilité articulaire (H. A.), a beaucoup nui à la prise en compte des plaintes des patients. Grahame(5) et Bravo(6) ont considérablement fait évoluer la description clinique chez les rhumatologues en l’enrichissant progressivement (douleurs, fatigue, troubles digestifs, dysautonomie, situations de handicap, etc.). La deuxième discipline médicale qui s’est investie dans ce syndrome est la génétique à partir de 1949. Beighton donnera son nom à un test d’hypermobilité en 9 points, imparfait, souvent mal appliqué et mal interprété ; il sert encore fréquemment, à l’instar de l’étirabilité, à éliminer un diagnostic pourtant évident devant la présence d’autres symptômes tout autant significatifs. Autour de Beighton et de son école, se met en place une classification basée sur les mutations des collagènes. Cette classification partie de 11 types, est réduite dans sa dernière version(7) à 6 et, en pratique courante, à 3 (classique, hypermobile, vasculaire). Elle préoccupe beaucoup les patients qui craignent d’être atteints du SED-vasculaire décrit comme létal avec le risque de rupture d’anévrysmes et de déchirures graves d’organes, ce qui est exceptionnel. Pour l’identifier, la mise en évidence d’une mutation du COL3A1 est considérée comme un argument fort, mais il est inconstant. Dans les critères de la classification de Villefranche, manquent la plupart des symptômes du SED. Les critères différentiels entre les divers « types » sont redondants et imprécis. En pratique, cette classification est inutilisable et à l’origine de bien des malentendus préjudiciables aux patients si on l’utilise pour refuser des prises en charge de soins, pourtant légitimes, par les médecins conseils et les Maisons départementales des personnes handicapées (MDPH). Il est donc nécessaire de substituer à cette classification, des critères diagnostiques de certitude, ce qui est aujourd’hui possible. Il apparaît clairement(8) que l’hypermobilité articulaire des rhumatologues et le syndrome d’Ehlers-Danlos hypermobile des généticiens sont une seule et même entité clinique qui relève des mêmes préventions et contre-indications face aux risques (iatrogènes surtout) et des mêmes traitements du syndrome proprioceptif. Cette unicité de la vision sémiologique de ce syndrome doit rapidement faire évoluer la situation médicale et sociale dans laquelle se trouvent ces patients, qui a fait dire à Rodney Grahame, au récent colloque international francophone de Créteil du 7 mars 2015 « Les traitements du syndrome d’Ehlers-Danlos » : « Malheureusement, les patients qui souffrent du SED ne sont pas bien pris en charge par la profession médicale. Ils ne sont ni écoutés, ni crus, ni bien diagnostiqués ni correctement traités ». Parmi les signes les plus solides pour établir le diagnostic, figurent des manifestations cutanées très évocatrices qui peuvent conduire à consulter en dermatologie ; c’est dire le rôle important des dermatologues dans le dépistage de ce syndrome. Diagnostic clinique de syndrome d'Ehler-Danlos Le diagnostic s’appuie aujourd’hui sur l’interrogatoire du patient et de sa famille et un examen clinique simple. Nous présentons ici les manifestations recueillies depuis l’observation de 2 500 patients sur 17 années de consultations spécialisées. Elles se rapprochent des nouvelles descriptions séméiologiques de la littérature médicale mondiale(9). Ce syndrome apparaît comme un désordre global de la proprioception (véritable sixième sens) intéressant la perception multisensorielle du corps externe, mais aussi du corps interne et des interfaces du corps avec son environnement spatial et social. Ce concept global permet une analyse, à la fois des symptômes et des hypothèses physiopathologiques qui guideront les choix thérapeutiques. • Les douleurs(10) – diffuses (j’ai mal partout), articulaires et périarticulaires, musculaires, abdominales, thoraciques, génitales (dysménorrhée), céphaliques (migraines souvent), cutanées (signe de l’étiquette : retrait des étiquettes des chemisiers) ; – permanentes, mais avec des crises qui peuvent être d’une très grande violence, elles résistent volontiers aux antalgiques, même puissants. Ces crises peuvent être la conséquence d’une activité physique, elles sont alors souvent décalées au lendemain, véritable « dette douloureuse ». • La fatigue est le symptôme le moins bien toléré ; imprévisible, elle est un obstacle sérieux à l’activité professionnelle et à l’insertion scolaire. • Les troubles du sommeilsont habituels et importants. • Les troubles proprioceptifs du contrôle du mouvement sont à l’origine de désordres articulaires avec des pseudoentorses, des subluxations ou luxations, mais aussi des diminutions ou des pertes transitoires de la commande volontaire et de la perception de tout un secteur corporel réalisant des tableaux de « pseudo-paralysies » transitoires mais déroutants. Ils sont responsables d’un signe très évocateur : le signe de la porte (heurt des encadrements) et de maladresses (« syndrome de Pierre Richard »). Il faut rapprocher des désordres moteurs ceux dont l’origine est une dystoniequi est fréquente, repérable par l’existence de contractions (« secousses ») involontaires. Elle joue un rôle important (douleurs, subluxations...) dans les difficultés fonctionnelles rencontrées par ces patients. • L’hypermobilitéest présente à condition de prendre en compte son expression depuis l’enfance (elle peut disparaître avec l’âge), les limitations par résistance musculaire douloureuse, les rétractions des muscles postérieurs des membres inférieurs (67,5 % des cas), et pas seulement par le test de Beighton (toucher la face antérieure de l’avant-bras avec le pouce en flexion forcée, extension du 5e doigt à 90°, recurvatum du coude à 10° au moins, recurvatum du genou à 10° au moins, toucher le sol avec la paume des mains) (figures 1 et 2). Figure 1. Genoux du SED : recurvatum et ecchymoses. Figure 2. Luxation des tendons stabilisateurs externes du pied par distension des coulisses fibreuses. • La fragilité de la peau constitue un élément objectif très important: elle est mince, douce et veloutée au contact, transparente, laissant bien voir les lacis veineux. Les rides sont discrètes ou absentes (les personnes avec un SED paraissent toujours plus jeunes). Les paupières peuvent être tombantes. Elle protège mal de l’électricité statique et les patients ressentent souvent des décharges électriques en ouvrant leur portière de voiture (« signe de la portière »), en prenant un caddy et au contact d’une autre personne. Sa minceur et la fragilité des vaisseaux attirent les moustiques, les puces, les araignées. Elle s’écorche facilement. Les vergetures sont fréquentes dans les deux sexes et parfois volumineuses. La cicatrisation est perturbée : lente, de mauvaise qualité, avec lâchages des sutures. Les chéloïdes sont rares, l’aspect papyracé est plus fréquent. L’étirabilité est inconstante et, le plus souvent, modérée. Elle doit être recherchée à la face, au cou, à l’avant-bras plutôt qu’au coude ou sur le dos de la main. C’est dans la peau qu’un grand nombre de perceptions nécessaires à un bon contrôle proprioceptif trouvent leur origine. Les modifications de l’élasticité, la minceur détériorent fortement leur qualité (figures 3 à 6). Figure 3. Vergetures et ecchymose. Figure 4. Fragilité de la peau qui se déchire facilement et ecchymoses. Figure 5. Signe de Miget (en référence à une illustration de la thèse de Miget). Hématome lors d’une ponction veineuse. Figure 6. Éruption psoriasiforme parfois observée dans le SED. • La dysautonomie, très bien décrite par Bravo, est constante : frilosité, sudations abondantes, extrémités froides avec pseudo-syndrome de Raynaud (signe de la chaussette : port de chaussettes la nuit), accélérations du rythme cardiaque, tension artérielle variable (le plus souvent basse), bouffées vasomotrices, malaises orthostatiques (POTS). • Les hémorragiesconstituent un excellent argument pour le diagnostic dans toutes les formes cliniques du SED. Elles doivent être bien connues du patient et de ses médecins pour éviter les accidents par traitement anticoagulant ou antiagrégant plaquettaire et les accidents hémorragiques lors de gestes chirurgicaux d’accouchements. Elles se manifestent par des gingivorragies, des épistaxis, des ménorragies/métrorragies, des saignements fibromateux, des ruptures hémorragiques de kystes de l’ovaire ou d’une grossesse extra-utérine. Elles sont responsables, au long cours, de déglobulisations et d’hyposidérémies. • Les manifestations respiratoiressont très fréquentes (souvent confondues avec l’asthme), il s’agit de dyspnée, notamment à la montée d’escaliers (« signe de l’escalier ») et de sensations de « blocages » inspiratoires, parfois lors de douleurs thoraciques, pouvant être confondues avec une crise d’asthme ou une embolie pulmonaire. • Les manifestations cognitives, telles que l’altération de la mémoire de travailet les troubles de l’attention, forment une part très importante, pas ou peu étudiée, des difficultés rencontrées chez ces patients. En revanche, l’intelligence est habituellement très développée, objectivable par les succès scolaires. Des manifestations comportementales, souvent observées chez l’enfant avec un SED, posent la question de la frontière avec la psychopathologie, notamment l’autisme. Des interférences existent, mais les confusions semblent plus fréquentes. • À côté de la symptomatologie décrite précédemment, s’ajoute un critère diagnostique qui a un poids considérable c’est le caractère familialchez les parents, les enfants, les frères ou sœurs, les cousins, signant le caractère génétique autosomique de cette pathologie avec une répartition non mendélienne. Le risque génétique est manifestement plus élevé que dans la transmission autosomique dominante habituelle avec une symptomatologie très variable dans une même famille, les formes frustes sont fréquentes et les tableaux retrouvés sont souvent incomplets ou discrets, difficiles à identifier. Autres manifestations cliniques • Gastro-intestinales : reflux gastro-œsophagien, constipation, ballonnements. • Bucco-dentaires avec un palais ogival, des problèmes d’articulé dentaire, rétractions gingivales, allongement du frein de la langue permettant de toucher le nez avec le bout de la langue (signe de Gorlin), fragilités et mobilités dentaires, inefficacité des anesthésiques locaux (figures 7 et 8). • ORL : hyperacousie, hypoacousie, voire surdité, acouphènes, hyperosmie, vertiges. • Ophtalmologiques : fatigue visuelle, myopie. • Gynécologiques et obstétricales : règles abondantes, accouchements difficiles. • Manifestations vésico-sphinctériennes : difficultés à uriner, envies pressantes, fuites. Figure 7. Signe de Gorlin, souvent absent : toucher le nez avec la langue. Figure 8. Implantations dentaires pathologiques. Rétractions gingivales. Quelques aspects dermatologiques particuliers Nous avons observé plusieurs manifestations cutanées non rapportées par les auteurs qui publient sur ce syndrome : prurit généralisé, sécheresse de la peau, modifications des phanères (alopécie, fragilités unguéales), éruptions eczématiformes ou psoriasiformes, aspects marbrés localisés ou diffus, zones de vitiligo plus ou moins étendues. Les tatouages ne sont pas altérés au fil des années, même si la mobilité cutanée est importante. En revanche, les piercings sont l’occasion de déboires tels que la déchirure d’un lobe d’oreille sous l’effet de la traction d’une boucle un peu pesante. Ils sont à déconseiller aussi bien au niveau de la peau que des muqueuses. La présence de nodules sous-cutanés, de masses ou de kyste est habituelle, correspondant probablement aux tumeurs molluscoïdes souvent mentionnées par les premiers descripteurs. Les examens complémentaires Les tests génétiques Ils ne sont contributifs au diagnostic que dans certaines formes rares du syndrome et seulement s’ils sont positifs. Ils restent l’apanage de consultations de génétique très spécialisées. Les biopsies cutanées ne sont pas nécessaires au diagnostic devant une clinique évocatrice. Elles sont mal supportées du fait de l’inefficacité des anesthésiques locaux et laissent des cicatrices inesthétiques et durablement douloureuses. Les électro myogrammes sont normaux. Les IRM articulaires, souvent prescrites, ne sont pas contributives, sauf par leur négativité, Elles peuvent être trompeuses (le pincement discal est habituel du fait de la souplesse du conjonctif). L’IRM cérébrale par tenseur de diffusion (séquences Flair et T2 étoile) Elle met en évidence des images très évocatrices du syndrome, notamment au niveau des noyaux lenticulaires qui permettent à l’imagiste entraîné d’évoquer le diagnostic. Les échographies de l’ensemble de l’arbre artériel À la recherche de dilatations, dissections ou anévrysmes artériels, elles sont systématiques et à répéter selon les résultats, et au minimum tous les 3 ans. L’échographie cardiaque peut déceler des ballonisations de valves, des fuites mitrales ou aortiques, sans aucune conséquence hémodynamique mais ce sont des éléments contributifs au diagnostic. Les radiographies du rachis dorso-lombaire Elles ne sont indiquées que lorsque l’examen du déroulement du rachis a décelé une voussure lombaire et/ou une gibbosité qui doivent être surveillées. Ces radiographies doivent être réalisées en position couchée, la radiographie du rachis en position debout peut donner de fausses images de « scolioses» par rotation de vertèbres qui sont hypermobiles. D’autres bilans peuvent compléter utilement l’évaluation Audiométrie, bilan orthoptique, tests posturologiques, échographies pelviennes, bilans urodynamiques, dosage de la vitamine D (diminuée de façon importante). La vitesse de sédimentation peut être augmentée, les CPK aussi, témoins de réactions inflammatoires peut-être secondaires à des microtraumatismes sur des tissus fragiles. Le fer sérique peut être bas, témoin de la déglobulisation par saignements chroniques. Histoire naturelle, évolution dans le temps Aujourd’hui, le diagnostic est trop tardif (21 ans en moyenne, chez les femmes, 15 ans chez les hommes) après l’apparition des premiers signes évocateurs), ce qui entraîne une errance thérapeutique toujours préjudiciable au patient et à sa famille. L’évolution de cet état de fragilité est imprévisible. La symptomatologie est plus sévère chez les femmes. L’espérance de vie, si on excepte les très rares formes à expression vasculaire majeure et la possibilité d’accident iatrogènes, n’est pas réduite. La qualité de vie avec de multiples situations de handicap, en revanche, est compromise. Certains facteurs agissent nettement sur la symptomatologie : l’état hormonal dans cette population à dominante féminine (influence de la puberté, des grossesses, des phases du cycle menstruel), la climatologie (effet négatif du temps froid et humide), les traumatismes posant des problèmes médico-légaux lors d’accidents de la voie publique, l’intervention chirurgicale sur une grosse articulation a parfois les mêmes effets négatifs qu’un traumatisme. Traitements Contrairement à une opinion trop souvent exprimée, Il existe des traitements efficaces du syndrome d’Ehlers-Danlos contre la proprioception, les douleurs et les manifestations digestives, bucco-dentaires, urinaires, etc. Les avancées les plus remarquables sont l’apport des orthèses (vêtements compressifs proprioceptifs surtout), de l’oxygénothérapie combinée ou non au percussionaire, des injections locales de lidocaïne et de la Dopa dans la dystonie. L’apport de la rééducation est essentiel dans ce syndrome où bouger reste la meilleure des thérapeutiques. Il faut être très prudent avec la chirurgie. • Contre les désordres proprioceptifs, recourir principalement aux orthèses : orthèses plantaires (appui rétrocapital médian, voûte de soutien, support sous-cuboïdien), vêtements compressifs proprioceptifs, dérivés des vêtements des brûlés avec des caractéristiques propres et spécialement adaptés au SED, ceinture lomboscapulaire avec trois points. Les colliers, les coudières, les genouillères, les orthèses des poignets et des doigts viennent compléter, au cas par cas, l’équipement orthétique (figures 9 et 10) tronc et du cou. Figure 9. Vêtements proprioceptifs compressifs issus d’une recherche à partir de ceux des brûlés. Figure 10. Utilisation d’une orthèse cruro-pédieuse articulée de marche. Le traitement orthétique des scolioses sévères, heureusement rares, se situe à part (figures 11 et 12). Figure 11. Coque moulée montée sur un siège mobile pour stabiliser le bassin et le tronc. Figure 12. Déshabillage par un chien d’assistance (difficulté majeure à mobiliser l’épaule). Les améliorations du syndrome proprioceptif postural par les verres prismatiques, l’orthoptie et le port d’orthèses dentaires sont des thérapeutiques prometteuses. La kinésithérapie et la kinébalnéothérapie proprioceptives, la rééducation des chaînes musculaires (Kabat), l’ergothérapie, la natation, le Tai Chi Chuan, l’équitation, la relaxation, la sophrologie, l’autohypnose, le biofeed back, le mouvement imaginé et l’imagerie guidée, le mouvement virtuel, le yoga, etc. Ce qui est essentiel c’est de maintenir une activité motrice et d’informer les systèmes de neurorégulation (pyramidal, extrapyramidal, neurovégétatif) de l’état corporel et de ses variations. L’activité sportive est un excellent moyen. Aucun sport n’est contre-indiqué, mais des adaptations peuvent être nécessaires. Dans l’enfance et l’adolescence, les performances sportives sont fréquentes. La chirurgie orthopédique a une place très limitée (épaules, mains) et est souvent (genoux, pieds, rachis...) à l’origine d’aggravations locales (douleurs, algodystrophies) et plus diffuses du syndrome. • La proprioception respiratoire est un point essentiel et implique une rééducation et une autorééducation respiratoire, diaphragmatique en particulier, le recours au chant, l’apport du percussionnairesont ici très utiles. Ce dernier a permis de libérer les patients des accidents de blocages, de fausses routes, de diminuer la fréquence des bronchites, des infections des voies aériennes supérieures et des pauses respiratoires angoissantes. Combiné à l’oxygène, il a aussi un effet sur la dyspnée souvent handicapante. L’oxygénothérapie, seule, régulière, à domicile et à l’école ou au travail, apparaît, aujourd’hui, comme l’un des traitements de base du SED, chez l’adulte comme chez l’enfant, par ses actions sur la fatigue, les douleurs (maux de tête surtout). • Contre les douleurs, il faut privilégier les traitements locaux : TENS, emplâtres de lidocaïne, injections locales (sur les points gâchettes musculaires, paratendineuses) de lidocaïne, très efficaces, aux effets étonnamment durables, gels anti-inflammatoires, agents physiques (ultrasons, chaleur surtout, mais aussi cryothérapie corps entier qui commence à donner des résultats). Les massages et les étirements par leurs effets décontracturants et proprioceptifs ont une place importante. Les orthèses ont un effet antidouleur souvent très net. Les antalgiques généraux doivent être utilisés avec circonspection, ils sont souvent mal ou très mal tolérés, ils conduisent parfois à des addictions pénibles. Nous proposons l’utilisation continue d’un myorelaxant (Baclofène®) et lors de crises seulement, du Tramadol® et surtout du Néopam®, qui apparaissent comme les plus efficaces et les mieux tolérés. Les anti-inflammatoires, sous protection gastrique, surtout après des douleurs post-traumatiques, peuvent être utiles. La L-carnitine est d’un apport quasi constant sur les douleurs, la fatigue, la constipation. Le thermalisme adapté à la fatigabilité et à la fragilité cutanée a montré son efficacité et constitue un excellent cadre pour l’éducation des patients. La dystonie est fréquente et joue un rôle dans la survenue des douleurs et du désordre locomoteur, parfois avec des crises spectaculaires de contractures musculaires. Le recours aux anti-parkinsoniens est ici très efficace, la toxine botulique a logiquement une place. • Contre la fatigue qui est très intimement mêlée aux phénomènes douloureux, les traitements précédents ont une efficacité certaine. Le sommeil est très souvent perturbé et peut être amélioré par la mélatonine et la luminothérapie. • D’autres traitements à visée gastro-intestinale, vésicosphinctérienne, gynécologique sont indiqués en fonction de la symptomatologie. Une supplémentation en vitamine D est quasiment systématique ; elle peut être nécessaire aussi pour le fer. Le traitement médicamenteux de la dysautonomie est difficile. • Les troubles cognitifs bénéficient avec succès des techniques d’orthophonie utilisées pour les traumatisés cérébraux légers (mémoire, attention, orientation). • Un accompagnement psychologique, voire psychiatrique, est très souvent nécessaire, impliquant la famille ; il est difficile chez les adolescents. La confusion entre le SED et un état psychopathologique est possible et la frontière n’est pas toujours facile à tracer. Conclusion La méconnaissance de la transmission familiale et la privation des thérapeutiques innovantes mises en place depuis quelques années sont un préjudice pour ces patients. Il est urgent de faire évoluer la situation dans laquelle se trouvent la plupart des patients avec un SED ainsi qu’en témoigne ce courriel reçu tout récemment : « Depuis 2010, j’ai une suspicion de Syndrome d’Ehlers-Danlos. En effet, j’ai énormément de symptômes. Cette suspicion a été évoquée dans un compte-rendu médical par le Docteur N. Cela fait 7 ans que je souffre, que l’on m’a diagnostiquée fibromyalgique par facilité étant donné qu’énormément de médecins ne connaissent pas le syndrome d’Ehlers-Danlos, d’autre part beaucoup de médecins se refusent à le diagnostiquer quand bien même ils le connaissent ». Cette situation doit changer.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :