Maladie de système, Médecine interne
Publié le 30 juin 2015Lecture 17 min
Cas particuliers d’hyperhidrose
A. JEANSON, C. GRAPINET, CHU de Besançon
L’hyperhidrose est un symptôme que l’on trouve dans certaines pathologies neurologiques telles que la maladie de Parkinson, la sclérose en plaques ou la syringomyélie, mais également dans d’autres pathologies plus rares.
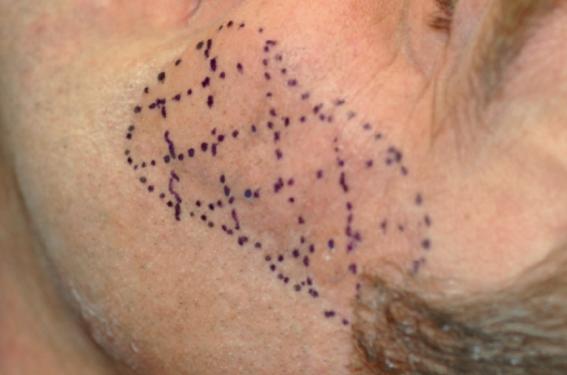
D.R. Test à l'iodine permettant de diagnostiquer un syndrome de Frey. Granulosis rubra nasi Exceptionnel, granulosis rubra nasi s’observe chez les garçons âgés de quelques mois ou années (avant 10 ans). Un érythème franc de la pointe du nez se couvre de vésiculettes de sueur eccrine. L’hypersudation peut précéder l’érythème de plusieurs années. Une acrocyanose peut être associée. Secondairement, joues et menton peuvent être atteints. La régression apparaît vers 3-4 ans ou à la puberté, avec télangiectasies et microkystes résiduels. Il n’y a pas de traitement efficace reconnu. Syringomyélie La syringomyélie est une maladie de la moelle épinière liée au développement d’une cavité en son centre qui tend à comprimer et à détruire progressivement la substance grise puis la substance blanche. Les causes de l’apparition de la cavité intramédullaire sont inconnues. Son évolution spontanée se fait vers un agrandissement avec altérations anatomiques de plus en plus importantes. Dans plus de 90 % des cas, la syringomyélie est associée à une malformation d’Arnold-Chiari : le bulbe et les tonsilles cérébelleuses (deux structures du tronc cérébral normalement contenues dans la boîte crânienne) se trouvent en aval du foramen magnum (ce qui peut entraîner leur compression). Les signes et symptômes de la maladie sont donc causés par une compression et une destruction des structures de la moelle à partir d’une cavité qui grandit à ses dépens. Une anesthésie thermo-algésique (le patient ne ressent plus le chaud et froid, ni la douleur), et une sensation subjective de brûlures fait partie d’un syndrome lésionnel. L’hypersudation résultante est souvent localisée. Syringobulbie Affection du bulbe, analogue, sur le plan anatomique, à la syringomyélie, caractérisée cliniquement par des troubles variables selon les noyaux bulbaires atteints. Neuropathie périphérique autonome et sensitive chez le diabétique La neuropathie diabétique est définie par l’atteinte du système nerveux périphérique (neuropathie périphérique) et du système nerveux végétatif (neuropathie végétative, neuropathie autonome ou dysautonomie). La neuropathie diabétique est classiquement la plus précoce des complications chroniques. Elle est extrêmement fréquente. La prévalence varie d’une étude à l’autre, de 5 à 60 % en fonction des auteurs. Ces différences sont surtout liées à la disparité des critères utilisés pour la définition de la neuropathie (examen clinique ou explorations neurophysiologiques). La gravité de cette complication est surtout liée aux conséquences cliniques qu’elle entraîne (troubles trophiques, douleurs neuropathiques, atteintes dysautonomiques sévères). Parmi ces troubles, on retrouve les manifestations sudorales, caractérisées par une absence de sudation au niveau des membres inférieurs, contrastant avec des épisodes d’hypersudation au niveau de la partie supérieure du corps et du tronc, déclenchés en particulier par la prise de repas. Ces manifestations peuvent être une source d’erreur dans le diagnostic des hypoglycémies nocturnes et entraîner des erreurs dans l’adaptation de traitement. Maladie de Parkinson On peut observer d’autres troubles, de type dysautonomiques, et notamment troubles de la sudation chez le Parkinsonien : – hypersudation ; – hypersécrétion sébacée. Ces dernières années, les résultats de la recherche, sur l’homme également, ont prouvé sans équivoque que dans le cadre de la maladie de Parkinson, les neurones du système dopaminergique (dans la substance noire) ne sont pas les seuls à mourir. D’autres systèmes de neurotransmetteurs sont également touchés. Ainsi, les neurones noradrénergiques et sérotoninergiques dépérissent également de manière anticipée – et ce, dès les stades précoces de la maladie. Ces troubles sont observés aussi bien dans les centres de régulation du tronc cérébral que dans les ganglions des nerfs périphériques. En résultent différents symptômes dits « non moteurs » du Parkinson – parmi lesquels le trouble de la sudation. En règle générale, ce dernier se manifeste d’une part par une sécrétion accrue de sébum par la peau, notamment dans la région du visage et du sillon sternal, et d’autre part par une tendance accrue à la sudation pour réguler la température en cas de chaleur (en particulier si le climat est humide). Étant donné qu’il s’agit d’une conséquence d’un trouble d’un circuit logique nerveux complexe, la thérapie s’avère délicate et aucun traitement médicamenteux ciblé n’est connu. On a cependant observé que la sudation est plus importante pendant les phases « off » et qu’elle accompagne parfois les fluctuations motrices. Cela signifie donc également qu’il conviendrait d’optimiser la motricité à l’aide des antiparkinsoniens traditionnels, dans l’espoir qu’ils exercent une influence positive sur la sudation par limitation des variations d’action et des phases « off ». Sclérose en plaques La sclérose en plaques, notée SEP, est la pathologie neurologique non traumatique du système nerveux central la plus fréquente chez les jeunes adultes. Elle se caractérise par une réaction inflammatoire développée contre la myéline du système nerveux central et détériore ainsi la qualité des influx nerveux. Elle touche 80 000 personnes en France, en majorité des jeunes adultes et des femmes. La SEP est la première cause de handicap non traumatique chez les jeunes adultes. Les phénomènes dysautonomiques sont fréquents lors de la sclérose en plaques, mais le plus souvent à des stades évolués. Une atteinte des voies centrales du système sympathique peut entraîner une hypersudation par un phénomène supposé de désinhibition synaptique. Syndrome de Shapiro Le syndrome de Shapiro, décrit dans les années 1960, est une cause rare d’hypothermie (température < 35 °C), responsable d’épisodes périodiques associant hypothermie sévère et hyperhidrose. Les hypothermies vues aux urgences sont habituellement accidentelles, métaboliques, toxiques ou iatrogènes, septiques, voire neurologiques (atteinte du système nerveux central) et cette cause inhabituelle (moins de 30 cas décrits dans la littérature internationale) peut poser des problèmes diagnostique et thérapeutique aux urgences et en réanimation. L’hypothermie périodique spontanée, ou syndrome d’hypersudation-hypothermie de Shapiro, semble liée à une atteinte directe des centres thermorégulateurs hypothalamiques, avec ou sans atrophie du corps calleux. Initialement décrits en association avec des anomalies morphologiques du corps calleux, les cas d’hypothermie périodique spontanée peuvent survenir en l’absence de toute anomalie structurale de l’hypothalamus, du corps calleux ou des lobes frontaux à l’imagerie cérébrale ; l’étiologie est inconnue et vraisemblablement multifactorielle, compte tenu de l’hétérogénéité des cas décrits. En revanche, sur un plan clinique, les crises sont stéréotypées avec hypersudation puis hypothermie et enfin réascension thermique. Durée (de quelques heures à quelques jours), fréquence (d’une crise par jour à des intervalles de plusieurs semaines) et intensité des crises sont variables touchant indifféremment les enfants et les adultes. Des crises d’hypersudation localisée sont également possibles (hémicorps, par exemple), mais respectent la même séquence d’apparition des symptômes, c’està-dire sueurs puis baisse de la température. Les anomalies biologiques parfois observées (élévation des transaminases, hyponatrémie, thrombopénie ou pancytopénie) ne sont pas pathognomoniques et pourraient être imputées à l’hypothermie quelle qu’en soit la cause. L’imagerie cérébrale (IRM notamment) est peu contributive, car elle peut être normale chez des sujets atteints et a contrario une agénésie du corps calleux est retrouvée chez 0,1 à 0,7 % des sujets non atteints de syndrome de Shapiro. L’utilisation de techniques d’imagerie métabolique et fonctionnelle (tomographie d’émission monophotonique avec amines marquées au 99 m Technetium) a récemment mis en évidence des anomalies de débit sanguin hypothalamique alors que l’IRM était négative. Le pronostic dépend de la profondeur de l’hypothermie même si aucun décès imputable n’est à déplorer ; l’évolution naturelle de la maladie conduit à un espacement des crises, voire à leur disparition. Dans l’état actuel des connaissances, le traitement est essentiellement symptomatique en fonction de la profondeur de l’hypothermie ; la clonidine a été proposée, mais aucune étude ne valide son utilisation. Sur un plan préventif, l’exposition au froid, de même que certains traitements psychotropes pourraient favoriser les crises. Syndrome de Frey Le syndrome de Frey se caractérise par une rougeur et une sudation du visage dans la zone d’innervation du nerf auriculotemporal. Les signes de ce syndrome surviennent lors de stimuli gustatifs dans les cas où le nerf auriculotemporal a été endommagé. Ce nerf, en compagnie des fibres sensorielles préauriculaires et temporales, transporte les fibres parasympathiques à la glande parotide ainsi que les fibres sympathiques vasomotrices et sécrétomotrices à la peau de la région préauriculaire. À la suite d’un abcès parotidien, un trauma, une chirurgie mandibulaire ou une parotidectomie, les fibres parasympathiques peuvent être lacérées. En tentant de rétablir l’innervation, ces fibres nerveuses peuvent se régénérer le long des fibres sympathiques créant ainsi une communication avec elles. À cause de ces connexions nerveuses aberrantes, lorsque la salivation est stimulée, les glandes sudoripares sont involontairement activées et les joues du patient deviennent rouges et humides. Plus de 40 % des patients qui ont eu une parotidectomie développent un syndrome de Frey en tant que complication de la chirurgie. Cette condition est rare durant l’enfance, mais peut être aperçue lors d’accouchement avec l’utilisation de forceps. Les manifestations du syndrome surviennent seulement lorsque l’enfant mange de la nourriture solide. Les parents pensent souvent à tort que l’enfant est allergique. De plus, le tiers des diabétiques ayant des neuro pathies développent une sudation lors de l’alimentation. Dans ce cas, les dommages nerveux surviennent à cause d’une ischémie chronique et d’attaques immunitaires. Certains phénomènes ressemblant au syndrome de Frey surviennent lors de chirurgies de la glande sous-mandibulaire (syndrome de la chorde tympanique) ou du nerf facial près du ganglion géniculé (syndrome lacrymatoire gustatif). En ce qui concerne le syndrome de la chorde tympanique, le menton et la peau sous le menton présentent de la sudation et de la rougeur. En ce qui concerne le syndrome lacrymatoire gustatif, la mastication cause une production abondante de larmes. Présentation clinique Les signes et symptômes du syndrome de Frey sont la sudation, la rougeur, la chaleur et parfois la douleur dans la région préauriculaire et temporale pendant la mastication. Ce syndrome se manifeste entre 2 mois et 2 ans après une chirurgie de la parotide. Lorsque la douleur est présente, elle est habituellement faible. Il est possible de détecter la présence de sueur à l’aide d’un test à l’iodine. Une solution de 1 % d’iodine est appliquée sur la région atteinte et elle est laissée à sécher. La solution est couverte d’une couche d’amidon. Lorsque le patient mange quelque chose, la sueur se mélange à l’iodine et cette dernière réagit avec l’amidon pour produire une couleur bleutée. Cela est considéré comme étant une technique diagnostique pour le syndrome de Frey. Traitement et pronostic La majorité des cas sont légers et ne nécessitent aucun traitement. De plus, 5 % des patients atteints remarquent une résolution spontanée de leurs symptômes. Approximativement, 5 % des diabétiques ayant des problèmes rénaux verront une amélioration considérable de leur syndrome de Frey à la suite d’une transplantation rénale. Une autre option thérapeutique peut être la lacération du nerf auriculotemporal ou du nerf glosso-pharyngien. Aussi, des injections d’atropine, des applications de crème à base de scopolamine ou l’utilisation d’agents antimuscariniques systémiques peuvent diminuer les symptômes de sudation ou de rougeur. Mais aujourd’hui, les meilleurs résultats sont obtenus par une dénervation chimique indolore obtenue par des injections de toxine botulinique. En 2 à 5 jours, 100 % des cas voient disparaître la sueur gustative pour une durée de l’ordre de 6 à 15 mois. Le risque de survenue du syndrome de Frey est grandement diminué si un lambeau de fascia temporo-pariétal est placé entre la glande parotide et la peau de la joue lors d’une parotidectomie. Cas clinique Nous avons eu le cas dans le service d’un patient de 45 ans qui souffrait d’hyperhidrose de la joue gauche lorsqu’il mangeait. Un diagnostic de Frey a été posé. Une injection de Botox® a été réalisée et le patient n’est pas revenu, une seule injection a donc été suffisante. Le test au Lugol® permettait de suivre l’évolution de la pathologie. Après traitement, le test montre une diminution significative de l’hyperhidrose. Syndrome de Riley-Day La dysautonomie familiale (DF) est une maladie héréditaire caractérisée par une perte des sensations et par une altération sévère de l’activité du système nerveux autonome entraînant des dysfonctionnements multisystémiques. La DF affecte presque exclusivement la population juive d’Europe de l’Est avec une incidence annuelle de 1 sur 3 600 naissances. Elle touche aussi bien les hommes que les femmes. Elle est présente dès la naissance et est progressive. Les symptômes initiaux (depuis la naissance jusqu’à l’âge de 3 ans) incluent des problèmes de déglutition, des pneumonies d’aspiration, une hypotonie, une instabilité thermique avec hypersudation et un retard du développement. À la naissance, il n’y a pas de dysmorphie évidente, mais une expression faciale caractéristique peut apparaître au cours du temps. Une cypho-scoliose sévère et une petite taille sont fréquentes. L’absence de larmes lors des pleurs est un des principaux signes de la maladie, mais il peut ne pas être immédiatement reconnu (l’absence de débordement lacrymal est normal jusqu’à environ 7 mois). Ainsi, en général, le dysfonctionnement autonomique est révélé en premier lieu par des difficultés d’alimentation dus à des troubles de la motricité digestive (coordination oropharyngée anormale, péristaltisme œsophagien anormal, vidange gastrique erratique, reflux gastroœsophagien et épisodes prolongés de vomissements appelés « crises dysautomiques »). Une maladie pulmonaire chronique (secondaire aux aspirations répétées), une maladie pulmonaire restrictive (due à une scoliose), une faiblesse musculaire et un dysfonctionnement des chimiorécepteurs (entraînant une réduction de la réponse à l’hypoxémie) sont fréquents. Une hypotension orthostatique sans tachycardie compensatoire est souvent rapportée. Les patients peuvent aussi présenter une hypertension épisodique en réponse à un stress émotionnel ou une douleur viscérale. Une modification de la personnalité peut aller de l’irritabilité et du repli sur soi à l’excitation généralisée. Des crises cycliques de fréquence quotidienne, hebdomadaire ou mensuelle se manifestent chez 40 % des patients et surviennent au réveil. La perception de la chaleur et de la douleur est diminuée dans presque tous les cas, mais n’est pas absente. La sensibilité aux vibrations est normale. Les réflexes tendineux profonds sont diminués chez la plupart des patients. Une absence de réponse axonale à l’injection intradermique d’histamine et une absence de papilles fongiformes sur la langue sont des signes caractéristiques de la DF. Sa transmission est autosomique récessive. La DF est due à des mutations du gène IKBKAP localisé sur le bras long du chromosome 9 (9q31). Le diagnostic repose sur la reconnaissance clinique des dysfonctionnements sensitifs et autonomes. Les « principaux » critères incluent une alacrymie, une absence de papilles fongiformes, une diminution des réflexes rotuliens et un test histaminique anormal. Le diagnostic est définitivement confirmé par une analyse moléculaire. Le diagnostic différentiel inclut les autres neuropathies héréditaires sensitives et autonomiques. Étant donné l’expression clinique très variable de la DF, la prise en charge doit être adaptée à chaque patient. Elle est symptomatique et s’attache principalement à traiter l’alacrymie, les dysfonctionnements gastrointestinaux et respiratoires et la labilité de la pression sanguine. Syndrome de Ross Le syndrome de Ross montre typiquement la triade composée de pupilles toniques, d’aréflexie et d’hyperhidrose unilatérale. À cause de l’hyperhidrose unilatérale, il se forme de manière compensatoire sur le côté opposé du corps une hyperhidrose (unilatérale) localisée. L’étiologie en est inconnue, probablement en rapport avec une dégénérescence des fibres végétatives cholinergiques postganglionnaires. Une hyperhidrose inhabituelle ou une fièvre inexpliquée représentent les motifs habituels de consultation des patients. L’examen clinique associé aux explorations électrophysiologiques met en évidence les éléments typiques d’un syndrome de Ross. Syndrome POEMS Le syndrome POEMS est défini par la présence d’une neuropathie périphérique (P), et d’autres manifestations paranéoplasiques, les plus fréquentes d’entre elles étant l’organomégalie (O), l’endocrinopathie (E), une anomalie des plasmocytes (M), les altérations cutanées (S pour skin), un papillœdème, un œdème, des épanchements, une ascite et une thrombocytose. Tous les patients auront au moins une lésion osseuse sclérosante ou une maladie de Castleman coexistante. Le pic d’incidence du syndrome POEMS se situe entre la 5e et la 6e décennie ; sa prévalence est inconnue. Il n’est pas nécessaire d’avoir tous les signes du syndrome pour en faire le diagnostic, et une reconnaissance précoce de la maladie est importante pour en réduire la morbidité. Il est souvent accompagné d’une hyperhidrose, de même que le syndrome de Gopalan (ou syndrome des pieds brûlants) et les tableaux de pachydermopériostose. Le syndrome POEMS est fréquemment confondu avec une polyneuropathie inflammatoire démyélinisante chronique. La confusion diagnostique pose problème du fait que les thérapeutiques efficaces pour les polyneuropathies démyélinisantes (gammaglobuline IV, plasma- phérèse, et azathioprine) ne le sont pas pour les patients atteints d’un syndrome POEMS. La cause du syndrome POEMS est inconnue. Les bases du traitement de ce syndrome incluent une radiothérapie, des corticoïdes et une thérapie par des agents alkylants, comprenant une chimiothérapie à forte dose avec transplantation des cellules sanguines souches circulantes. Insensibilité congénitale à la douleur et hyperhidrose Bowsher et coll. décrivent, chez deux individus non apparentés, une nouvelle forme de neuropathie sensorielle caractérisée par une faible sensibilité à la douleur, au toucher et à la température ainsi que par une hyperhidrose. L’analyse des terminaisons nerveuses chez l’un des sujets révèle le maintien d’une innervation au niveau des vaisseaux sanguins et des glandes sudoripares de la peau, mais l’absence de pratiquement tous les autres types de terminaisons, ce qui pourrait être à l’origine de la sensibilité cutanée résiduelle observée. Hyperhidrose et agénésie du corps calleux L’agénésie du corps calleux est la plus fréquente des malformations cérébrales. Elle peut s’accompagner d’épilepsie focale à type de crises d’hypersudation. Son traitement est symptomatique. Causes médicamenteuses d’hypothermie-hypersudation (Shapiro-like) Plusieurs cas d’hypothermie médicamenteuse ont été décrits avec notamment des médicaments à usage fréquent comme l’aspirine, la metformine, les antihypertenseurs (amlodipine, ramipril, furosémide) ou encore certains neuroleptiques (clopixol, cyamémazine). Deux cas d’hypothermie secondaire à la prise d’olmésartan médoxomil ont été rapportés à la Food and Drug Administration (FDA). Ceci soulève l’intérêt de consulter les banques internationales de pharmacovigilance. Le syndrome de Shapiro est habituellement défini par la triade hypothermie récurrente, hyperhidrose et agénésie du corps calleux. Dans près de la moitié des cas, la notion d’un traumatisme crânien, de symptômes neuro psychiatriques, ou de prise médicamenteuse est retrouvée et suggère que des lésions ou dysfonctions subtiles des centres de la thermorégulation sont responsables d’accès d’hypo thermie, même lorsqu’aucune anomalie de l’imagerie encéphalique n’est mise en évidence.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :