Publié le 10 juin 2024Lecture 13 min
Mise au point sur les maladies bulleuses auto-immunes
Florine GUERROIS, service de dermatologie du Dr Gaudron, GHEF, site de Marne-la-Vallée-Jossigny
Les maladies bulleuses auto-immunes (MBAI) se caractérisent par la présence de lésions bulleuses et siègent à différents niveaux de l’épiderme. L’histologie doit porter sur une bulle récente, et l’immunofluorescence directe (IFD) doit être réalisé en peau saine péribulleuse ou en peau inflammatoire mais non décollée. Ce résumé détaillera les maladies bulleuses par décollement sous-épidermique, les maladies bulleuses par décollement intra-épidermique et enfin les soins de bouche nécessaires.
LES MALADIES BULLEUSES PAR DÉCOLLEMENT SOUS-ÉPIDERMIQUE
Les maladies bulleuses par décollement sous-épidermique (figures 1A et 1B) se caractérisent par la présence d’autoanticorps qui se fixent sur des protéines de structure de la membrane basale. La pemphigoïde bulleuse est la plus fréquente des MBAI (66 % des cas). La pemphigoïde gravidique est une forme touchant la femme enceinte. Les autres MBAI sont moins fréquentes (34 % des cas). On parle de pemphigoïde des muqueuses dès que l’atteinte muqueuse est prédominante. Par exemple, les lésions peuvent évoquer une maladie à IgA linéaire (lésions regroupées en rosette), avec de nombreuses lésions muqueuses. On parlera alors de pemphigoïde des muqueuses à type IgA linéaire.
Figure 1A. Résumé des maladies bulleuses par décollement sous épidermique.
Figure 1B. Schéma simplifié de l’hémidesmosome et de la jonction dermo-épidermique.
L’hémidesmosome est composé de la protéine BP230 et BP180 (partie NC16A), de la plectine et de l’intégrine α6β. La partie terminale COOH de la protéine BP180 descend dans la lamina densa. Les laminines 322 et g1 se trouvent à la jonction des deux laminas. Le collagène VII est ancré dans la zone fibrillaire du derme.
Pemphigoïde bulleuse
La pemphigoïde bulleuse (PB) est la dermatose bulleuse auto-immune la plus fréquente en France. Son incidence augmente depuis les dix dernières années.
• Clinique et diagnostic
Le diagnostic repose sur l’association de plusieurs critères résumés dans le tableau 1(1). À noter qu’il existe des formes atypiques (cf. tableau 1*) : bulles localisées sur les régions palmoplantaires (pemphigoïde dyshidrosiforme), bulles localisées sur un segment cutané (figure 3), lésions vésiculeuses, lésions végétantes des grands plis, lésions à type de prurigo nodulaire, etc. À noter aussi que devant une éruption bulleuse du sujet âgé (cf. tableau 1**), la valeur prédictive positive du dépôt linéaire d’IgG et/ou de C3 est de 97 % pour une pemphigoïde bulleuse si trois des quatre critères suivants sont réunis(2) :
– âge > 70 ans ;
– absence de lésion muqueuse ;
– absence de prédominance des lésions sur la tête, le cou ou la moitié supérieure du corps ;
– absence d’évolution cicatricielle des lésions cutanées.
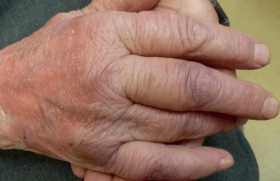
Figure 2. Pemphigoïde bulleuse. Placards urticariens symétriques des membres inférieurs, recouverts de bulles tendues et érosions post-bulleuses.
Figure 3. Pemphigoïde bulleuse. Larges érosions post-bulleuses sur la face d’extension d’une cuisse.
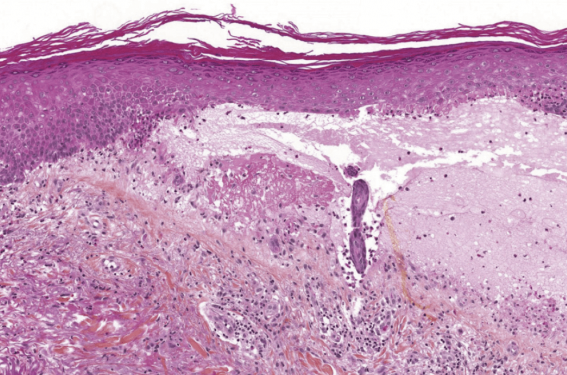
Figure 4. Pemphigoïde bulleuse. Décollement au niveau de la jonction dermo-épidermique avec présence de polynucléaires éosinophiles.
• Thérapeutique
L’âge, l’état général du patient et ses comorbidités sont des critères importants à prendre en compte dans la discussion thérapeutique. La mortalité est d’environ 30 % à 1 an, souvent corrélée au mauvais état général, au grand âge et aux comorbidités neurologiques du patient(3). Il est important de rechercher une dénutrition, des surinfections cutanées et de surveiller la tension artérielle et la glycémie chez les patients diabétiques. Le régime sans sel n’est pas nécessaire. Une supplémentation vitamino-calcique est conseillée mais n’a pas fait l’objet d’études. Les soins locaux sont indispensables et doivent clairement être expliqués aux personnels paramédicaux et aux patients. Il faut compter le nombre de bulles, les rapporter dans un carnet de suivi, et les percer tout en conservant leurs toits, puis tamponner le toit avec un antiseptique (type chlorhexidine aqueuse). Il faut appliquer un pansement gras sur les érosions et éviter l’utilisation d’adhésifs. Les traitements de 1re et 2e intention sont détaillés dans le tableau 2 et dans la figure 5(4,5).
Figure 5. Schéma récapitulatif des traitements, s’inspirant des PNDS actualisé(8).
Pemphigoïde gravidique
La pemphigoïde gravidique (PG) est une forme de pemphigoïde bulleuse, survenant au cours de la grossesse (généralement au 3e trimestre de gestation) ou en postpartum. Sa prévalence est estimée entre 1/1 700 et 1/50 000 grossesses. La protéine BP180 est exprimée, dès le 1er trimestre de la grossesse, dans les cellules cytotrophoblastiques et syncytiotrophoblastiques du placenta et dans les cellules épithéliales de la membrane amniotique. L’auto-immunisation pourrait survenir par perte de tolérance vis-à-vis de cet antigène. Par la suite, il y a une réponse immunitaire croisée contre la peau.
• Clinique et diagnostic
La clinique est similaire à la PB et les lésions débutent en région périombilicale. Les résultats des examens complémentaires sont similaires à ceux de la PB. Il y a plus de complications foetales (hypotrophie, prématurité) si la PG débute tôt dans la grossesse, est pluribulleuse et si le taux d’anticorps anti-BP180 est élevé au diagnostic. Le suivi obstétrical doit alors être rapproché. À noter, la PG récidive lors de grossesses ultérieures, souvent de façon plus précoce et plus sévère.
• Thérapeutique
Sur le plan thérapeutique, il est nécessaire de calmer le prurit et de supprimer les signes cutanés, en évitant toute complication locale (comme les surinfections) et systémique (complication iatrogène). En 1re intention, dans les PG modérées, les dermocorticoïdes de classe très forte sont efficaces. La corticothérapie générale est réservée aux formes sévères de la maladie.
Pemphigoïde cicatricielle
Les patients atteints de pemphigoïde cicatricielle (PC) sont plus jeunes que dans la PB.
• Clinique et diagnostic
Sur le plan clinique, l’atteinte cutanée est observée dans 25 à 40 % des cas, à type de bulles tendues en peau saine ou érythémateuse, évoluant vers des cicatrices atrophiques ou des grains de milium. L’atteinte du chef est prédominante. Toutes les muqueuses peuvent être atteintes, telles que :
– la muqueuse buccale (89 à 90 % des cas) : gingivite érosive, atteinte du palais, de la langue et des gencives ;
– la muqueuse conjonctivale (50 à 70 % des cas) : conjonctivite érythémateuse (stade 1) puis conjonctivite synéchiante avec symblépharon (stade 2). Il existe un risque de cécité par opacification cornéenne (stade 3) ;
– La muqueuse génitale (15 % des cas) : balanite ou vulvite bulleuse et/ou érosive et/ou synéchiante ;
– la muqueuse pharyngolaryngée (10 à 30 % des cas) : odynophagie, dysphonie avec sténoses laryngées graves ;
– la muqueuse œsophagienne (rare) : risque de sténose œsophagienne.
La gravité de la PC dépend des atteintes muqueuses (atteinte oculaire, laryngée ou œsophagienne). L’histologie, l’IFD, le test ELISA et l’IFI standard ne permettent pas de faire la différence avec une PB. L’IFI sur peau clivée retrouve une fixation des anticorps sur le versant épidermique ou dermique de la zone de clivage dermo-épithélial (figure 6).
Figure 6. Pemphigoïde cicatricielle. Biopsie d’une zone cutanée prébulleuse. Ébauche de décollement à la jonction dermo-épidermique.
• Thérapeutique
Sur le plan thérapeutique, la dapsone (50 à 100 mg/jour) est le traitement de 1re intention dans les formes de sévérité modérée. Elle est surtout efficace sur les lésions buccales ou cutanées et, à un degré moindre, sur l’atteinte oculaire. Avant de prescrire la dapsone, il faut éliminer un déficit en glucose-6-phosphate déshydrogénase (G6PD). Localement, des dermocorticoïdes peuvent être prescrits pour l’atteinte buccale et cutanée, et la ciclosporine pour l’atteinte oculaire. La corticothérapie générale est d’efficacité très inconstante. Les immunosuppresseurs, en particulier le cyclophosphamide (0,5 à 2 mg/kg/jour), ou le mycophénolate mofétil (1,5 à 3 g/jour) constituent le traitement de choix en cas d’atteinte oculaire. Le rituximab (anticorps anti-CD20) et les IgIV sont utilisés dans les atteintes oculaires sévères ou les atteintes réfractaires. L’efficacité s’observe surtout sur la composante inflammatoire oculaire et sur l’atteinte endobuccale après 4 semaines de traitement en moyenne.
Pemphigoïde à p200
Dans la pemphigoïde à p200, les anticorps réagissent avec la protéine dermique de 200 kDa correspondant à la chaîne gamma 1 des laminines. Une association avec le psoriasis a été remarquée, s’expliquant par le relargage de métalloprotéinases matricielles (MMP), dont la MMP9 qui dégrade la laminine gamma 1(9).
• Clinique et diagnostic
La pemphigoïde à p200 est caractérisée par un polymorphisme lésionnel se rapprochant de la PB et de la PC, avec la présence de lésions bulleuses tendues sur placard eczématiforme et/ou urticarien, une atteinte muqueuse (génitale et/ou buccale) et une évolution cicatricielle des lésions (grains de milium). Les explorations habituelles ne permettent pas de distinguer les 3 entités. On note toutefois à l’histologie que la bulle sous-épidermique s’accompagne d’un infiltrat dermique constitué de neutrophiles, formant parfois des micro-abcès au sommet des papilles dermiques. Le diagnostic est suspecté grâce à l’IFI sur peau clivée retrouvant un marquage du versant dermique et confirmé par l’immunoblot (réactivité avec la protéine dermique de 200 kDa).
• Thérapeutique
La prise en charge thérapeutique est hétérogène en l’absence de consensus. La corticothérapie locale très forte, la corticothérapie générale et la dapsone sont les traitements les plus souvent utilisés(10).
Dermatose à IgA linéaire
Dans la population pédiatrique, la dermatose à IgA linéaire est connue sous le nom de maladie bulleuse chronique de l’enfance (CBDC). Il s’agit de la plus fréquente des maladies bulleuses sous-épidermiques de l’enfance. Chez l’adulte, elle atteint les adultes d’âge moyen.
• Clinique et diagnostic
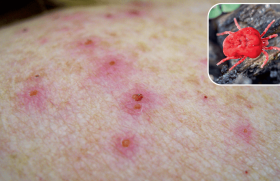
L’éruption cutanée est polymorphe : les bulles sont de taille variable se groupent en rosettes, en peau saine ou érythémateuse (figures 7 et 8). Le prurit est inconstant et la guérison est sans cicatrice. Dans 20 à 30 % des cas, on retrouve une atteinte muqueuse. Plusieurs cas d’induction médicamenteuse ont été rapportés, notamment avec la vancomycine, les inhibiteurs de l’enzyme de conversion, les anti-inflammatoires non stéroïdiens, les antiépileptiques (carbamazépine, lithium, vigabatrine). Il faut rechercher les maladies associées, telles que les maladies inflammatoires chroniques du tube digestif (rectocolite hémorragique, maladie de Crohn ou colite lymphocytaire), une hémopathie (les lymphomes non hodgkiniens et hodgkiniens et hémopathies lymphoplasmocytaires) et les maladies dysimmunitaires (connectivites, endocrinopathies). L’IFD met en évidence des dépôts d’IgA le long de la zone de la membrane basale. Dans 50 % des cas, des anticorps antimembranes basales de l’épiderme de classe IgA sont retrouvés en IFI.
Figure 7. Dermatose à IgA linéaire. Lésions bulleuses réparties en rosette au niveau de l’abdomen et du dos.
Figure 8. Dermatose à IgA linéaire. Décollement bulleux à la jonction entre l’épiderme et le derme avec de nombreux polynucléaires éosinophiles.
• Thérapeutique
Sur le plan thérapeutique, la dapsone (50 à 100 mg/jour) est le traitement le plus couramment utilisé. D’autres traitements peuvent être proposés, tels que les dermocorticoïdes, les cyclines, le corticothérapie générale de 0,5 à 1 mg/kg/jour, le rituximab, le méthotrexate et les IgIV.
Épidermolyse bulleuse acquise
L’épidermolyse bulleuse acquise (EBA) est une maladie rare, survenant souvent entre 40 et 50 ans. Elle est due à des autoanticorps ciblant le collagène de type VII, un composant crucial de l’ancrage des fibrilles dans la jonction dermo-épidermique.
• Clinique et diagnostic
Cliniquement, on décrit plusieurs formes d’EBA(11) :
– une forme chronique mécanobulleuse, souvent acrale : on retrouve des lésions bulleuses et des érosions sur les zones de frottement (mains, coudes, genoux, talons), secondaire à des traumatismes minimes, et une fragilité cutanée. L’évolution se fait vers une cicatrisation atrophique et des grains de milium. Des atteintes muqueuses (notamment la muqueuse buccale), des ongles dystrophiques et une atteinte du cuir chevelu évoluant vers une alopécie cicatricielle sont possibles ;
– une forme inflammatoire : cette forme prédomine sur le tronc et la partie proximale des membres ;
– la forme à type de pemphigoïde des muqueuses : atteinte possible de toutes les muqueuses malpighiennes ;
– la forme Brunsting-Perry-like : présence de lésions uniquement au niveau du visage et du cuir chevelu avec cicatrisation atrophique. Il n’y a pas d’atteinte muqueuse.
L’EBA a été associée à diverses maladies systémiques, notamment les MICI, la thyroïdite, la polyarthrite rhumatoïde, l’infection par l’hépatite C et le diabète sucré. L’histologie et l’IFD sont semblables à celles de la PB. L’IFI sur peau clivée retrouve un marquage sur le versant dermique du décollement dermoépidermique. En ELISA, on retrouve des anticorps anticollagène VII. On peut aussi utiliser le Biochip collagène 7.
• Thérapeutique
Des conseils sur les stratégies de prévention des traumatismes cutanés sont indispensables pour les patients, tels que limiter l’exposition aux savons agressifs et à l’eau chaude, s’abstenir de frotter vigoureusement la peau et éviter une exposition prolongée au soleil. La prise en charge thérapeutique n’est pas consensuelle(12). Dans les formes cutanées limitées, sans atteinte muqueuse, la corticothérapie locale, la colchicine, la salazopyrine et la dapsone sont privilégiées. Dans les formes cutanées diffuses ou avec une atteinte muqueuse sévère, on peut utiliser la ciclosporine, les IgIV et le rituximab.
Dermatite herpétiforme
La dermatite herpétiforme (DH) est rare. L’âge moyen au diagnostic est de 40 ans et elle est associée à la maladie cœliaque.
• Clinique et diagnostic
Elle est caractérisée par une éruption papulo-vésiculeuse symétrique, très prurigineuse, touchant la face d’extension des membres et les fesses. L’histologie met en évidence un infiltrat dermique superficiel, riche en polynucléaires neutrophiles réalisant des micro-abcès au sommet des papilles avec ébauche de décollement dermo-épidermique. L’IFD re trou ve des dépôts granuleux de C3 au sommet des papilles dermiques. Les examens immunosérologiques sont importants pour étayer le diagnostic :
– les anticorps anti-IgA antitransglutaminase épidermique ;
– le taux IgA sérique ;
– les anticorps IgA anti-transglutaminase tissulaire et anticorps IgA et IgG anti-peptide déamidé de la gliadine ;
– l’exploration digestive à la re cherche d’une maladie cœliaque doit être proposée.
• Thérapeutique
La dapsone (50 à 100 mg/jour) reste le traitement de premier choix(13). Elle permet une réduction du prurit et des lésions cutanées mais est inefficace sur l’atteinte digestive. Seul un régime sans gluten strict permet une action sur l’atteinte digestive et le taux des anticorps. Si le régime est bien suivi, la posologie de la dapsone peut être diminuée au bout de 6 à 12 mois, et un arrêt complet est envisageable dans une moyenne de 28 mois.
LES MALADIES BULLEUSES PAR DÉCOLLEMENT INTRA-ÉPIDERMIQUE
Les maladies bulleuses par décollement intra-épidermique se caractérisent par la présence d’autoanticorps ciblant les protéines des desmosomes, permettant la liaison entre les kératinocytes. Les protéines desmogléine (Dsg) contiennent cinq domaines extracellulaires, un seul domaine transmembranaire et un domaine cytoplasmique contenant des sites de liaison à la plakoglobine et à la plakophiline. L’envoplakine est principalement exprimée dans les kératinocytes différenciés alors que la périplakine est présente dans tout l’épiderme. Ces deux plakines permettent l’ancrage des filaments intermédiaires de kératine aux desmosomes (figure 9).
Figure 9. Schéma simplifié des protéines présentes entre deux kératinocytes.
Pemphigus (superficiel et profond)
Le pemphigus résulte de la perte de cohésion interkératinocytes par la présence d’anticorps antidesmogléine (Dsg) 1 et/ou 3.
• Clinique et diagnostic
Le diagnostic repose sur l’association de plusieurs critères(14) résumés dans le tableau 3. Le pemphigus est associé à d’autres maladies auto-immunes, le psoriasis, les troubles neurologiques et psychiatriques.
Figure 10. Pemphigus profond. A. Bulles en peau saine et érosion post-bulleuse péri-ombilicale. Marquage de la zone pour réaliser l’histologie (sur une bulle) et l’IFD (en peau péribulleuse). B. Atteinte du palais dur et mou et des lèvres.
Figure 11. Pemphigus superficiel. Décollement bulleux intra-épidermique avec acantholyse.
Figure 12. Pemphigus profond. Décollement bulleux intra-épidermique avec acantholyse.
• Thérapeutique
La prise en charge thérapeutique dépend de la sévérité de la maladie(15) (tableau 4). Durant le suivi thérapeutique, le dosage des anticorps circulants se réalise au diagnostic, tous les 6 mois ou en cas de rechute. La décroissance sera d’autant plus lente qu’il persistera un taux résiduel d’anticorps anti- substance intercellulaire ou un taux d’anticorps antidesmogléines > 50 UA/mL. La durée du traitement est en moyenne de 2 à 3 ans. L’arrêt du traitement peut être proposé chez un patient en rémission clinique sous faibles doses de corticoïdes per os (prednisone ou équivalent ≤ 5 mg/jour) ou de faibles doses d’immunosuppresseurs, d’autant plus que les anticorps sont négatifs.
Pemphigus paranéoplasique
Le pemphigus paranéoplasique est une maladie auto-immune rare (5 % des pemphigus) secondaire à une néoplasie (hémopathie : lymphomes B non hodgkiniens, leucémies lymphoïdes chroniques B, maladie de Castleman, thymome ou cancer solide : langue, poumon, estomac, côlon).
• Clinique et diagnostic
La clinique est polymorphe : présence de plaques lichénoïdes, de décollements cutanés, de lésions muqueuses érosives sévères. Il faut savoir y penser devant des lésions lichénoïdes atypiques et une atteinte muqueuse isolée et sévère, en particulier de la muqueuse buccale. En IFD, on peut retrouver un double marquage (marquage en résille et de la JDE). Les anticorps ciblent principalement la desmogléine 3 et les protéines plakine (envoplakine et périplakine).
• Thérapeutique
Le traitement comprend la corticothérapie générale +/- associée à azathioprine ou mycophénolate, anti-CD20 (rituximab), IgIV. Pemphigus néonatal Le pemphigus néonatal est une maladie transitoire chez les nouveau-nés, causée par le passage transplacentaire d’autoanticorps d’une mère atteinte de pemphigus. Les lésions cutanées sont présentes chez environ la moitié des nouveau-nés et disparaissent spontanément ou avec des corticostéroïdes topiques légers en 1 à 4 semaines.
LES SOINS DE LA MUQUEUSE BUCCALE
Les atteintes de la muqueuse buccale nécessitent une prise en charge médicale adaptée. Les patients ayant un dentier doivent le retirer tant que les lésions buccales ne sont pas contrôlées voire cicatrisées.
Hygiène dentaire
Une hygiène bucco-dentaire quotidienne est importante à maintenir chez les patients. Elle diffère en fonction de la sévérité de l’atteinte muqueuse(1) (tableau 5).
Corticothérapie locale
Pour les lésions buccales accessibles, une corticothérapie locale est recommandée. Il en existe différentes formes(1) :
– préparation magistrale contenant une corticothérapie locale (clobétasol) associée à de l’Orabase® à appliquer directement sur les lésions ;
– clobétasol gel à appliquer directement sur les lésions ;
– des bains de bouche avec prednisolone effervescent (un comprimé de 20 mg à faire fondre dans un fond d’eau, gargariser la solution pendant quelques minutes, puis recracher), 3 fois/jour ;
– des comprimés de Buccobet® à faire fondre au niveau des joues ;
– des sprays de corticoïde.
En conclusion
Les maladies bulleuses auto-immunes diffèrent par le mécanisme de formation des bulles, la clinique et le traitement. Il est important de savoir les reconnaître et de les prendre en charge rapidement pour diminuer le risque de mortalité (comme dans la pemphigoïde bulleuse), de limiter la rançon cicatricielle (comme dans les pemphigoïdes des muqueuses, notamment l’atteinte oculaire et œsophagienne) et de rechercher une cause néoplasique (comme dans le pemphigus paranéoplasique). Dans le cas des atteintes muqueuses, les soins de bouche sont importants à continuer, malgré les douleurs, pour limiter l’inflammation locale.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :