Publié le 21 jan 2021Lecture 3 min
Conduite à tenir devant un syndrome de sweet de l’enfant
H. SAHEL, Algérie

Le syndrome de sweet (SS) est une dermatose neutrophilique très rare chez l’enfant, réalisant entre 5 % et 8 % de tous les cas. L’âge de début moyen est de cinq ans. Les deux sexes sont atteints à égalité ; cependant avant l’âge de trois ans, il existe une légère prédominance masculine.
DIAGNOSTIC
Le tableau clinique et les anomalies histologiques sont identiques à ceux de l’adulte.
Le début est brutal, précédé parfois par des prodromes à type d’infections respiratoires ou digestives, puis des plaques apparaissent progressivement.
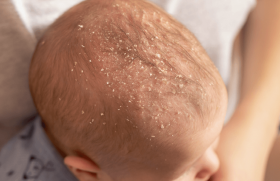
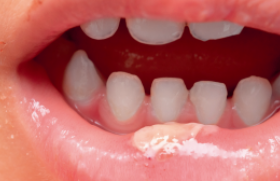
Elles sont érythémateuses, œdémateuses bien limitées, à surface mamelonnée, chaudes et douloureuses (figures 1 et 2).
Ces lésions évoluent dans un contexte fébrile avec malaise et myalgies. Elles siègent d’une façon symétrique sur le visage, le tronc, les extrémités et le cou.
Elles sont associées à un syndrome inflammatoire biologique avec une hyperleucocytose à polynucléaires neutrophiles (PN), une VS accélérée et une CRP élevée.
Le diagnostic est confirmé par l’étude histologique de la biopsie cutanée qui retrouve un infiltrat massif à PN dermique superficiel périvasculaire, sans signes d’une vasculite.
L’évolution du SS se fait vers la régression spontanée des lésions en 4 à 6 semaines, sans cicatrice. Rarement, elles laissent place à des lésions de type cutis laxa, fréquemment associées à des complications cardiaques qui peuvent entraîner le décès.
Des récidives peuvent se voir chez un tiers des patients, particulièrement dans le cas des SS associés à une néoplasie.
ASSOCIATIONS
Le SS peut être idiopathique/classique ou secondaire (para-inflammatoire ou paranéoplasique). Sont inclues dans les pathologies para-inflammatoires les maladies auto-immunes et les déficits immunitaires, et dans les pathologies paranéoplasiques les hémopathies essentiellement.
Alors que chez l’adulte l’association la plus fréquente est la pathologie néoplasique, chez l’enfant le SS est plutôt associé à des infections.
BILAN DE SURVEILLANCE
Il n’y a pas de consensus concernant le bilan biologique à réaliser au cours du suivi des enfants avec un SS. Certains préconisent de demander dans le bilan initial une NFS complète avec un frottis sanguin à la recherche d’une hémopathie. Elle doit être répétée jusqu’à la disparition de l’hyperleucocytose. Une biopsie de la moelle osseuse est nécessaire en cas de persistance de l’hyperleucocytose, d’une anémie ou d’une thrombopénie. D’autres préconisent de réaliser une échocardiographie chez tout enfant avec un SS associé à une cutis laxa.
Une exploration de la fonction de l’immunité humorale et cellulaire doit être demandée initialement, et répétée en cas d’infections à répétition à la recherche d’un déficit immunitaire.
Du fait de l’association de certains SS à une ostéomyélite focale chronique et récidivante, ces auteurs préconisent de réaliser un examen clinique musculo-squelettique avec des radiographies osseuses, à compléter si nécessaire par une IRM osseuse (tableaux 1 et 2).
TRAITEMENT
Le traitement de première intention du SS fait appel aux corticoïdes à la dose de 0,5 à 2 mg/kg/j.
En cas de récidives ou de résistance aux corticoïdes, d’autres alternatives thérapeutiques peuvent être utilisées, mais n’ayant pas fait la preuve de leur supériorité en termes d’efficacité par rapport aux corticoïdes. Il s’agit de la dapsone, de l’iodure de potassium, de la colchicine, et de l’indométacine.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :