Génodermatoses
Publié le 13 avr 2010Lecture 7 min
Porokératose linéaire de Mibelli associée à une porokératose en plaques
I. TADLAOUI, J. BOUHLLAB, L. BERBICH, B. HASSAM, Service de dermatologie, CHU Ibn Sina, Rabat, Maroc
Décrites la première fois en 1893 par Mibelli (1), les porokératoses représentent un groupe de dermatoses peu fréquentes, d’étiologie inconnue, de transmission autosomique dominante à pénétrance variable, qui répondent à un trouble de la kératinisation de l’épiderme. Cliniquement, les lésions peuvent être uniques ou multiples, petites ou de grande taille, à tendance atrophique ou hyperplasique, hyper- ou hypopigmentées, mais elles ont en commun une bordure périphérique surélevée et kératosique donnant un aspect caractéristique en histologie : lamelle cornoïde. Ces variations ont été à l’origine de différentes classifications.
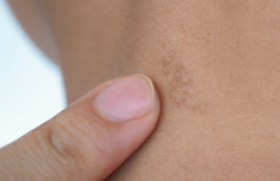
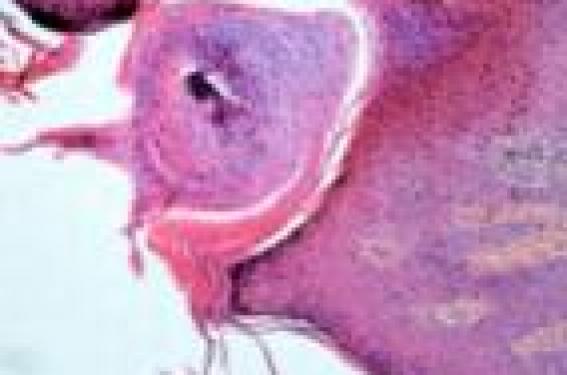
Observation Un patient de 17 ans, sans antécédents particuliers, issu de parents consanguins de deuxième degré, sans notion de cas similaire dans la famille, a consulté pour des lésions érythémato-squameuses et kératosiques de la face dorsale de la main droite et de l’avantbras homolatéral. Le début de la symptomatologie remontait à l’âge de 2 ans, avec l’apparition d’une lésion érythémato-squameuse, de petite taille, en regard de la face dorsale de l’avant-bras droit, indolore et non prurigineuse, évoluant d’un seul tenant et s’étendant progressivement et très lentement. Depuis 4 ans, est apparue une autre plaque linéaire sur la face dorsale de la main droite. Le tout évoluant dans un contexte de conservation de l’état général. À l’examen, on notait la présence d’un placard verruqueux hyperkératosique, unilatéral et linéaire, de contours réguliers, à centre atrophique et à bordure surélevée et hyperplasique au niveau de la face dorsale de la main droite. Une deuxième plaque érythémato-squameuse hyperkératosique siège en regard de l’avant-bras homolatéral. Le reste de l’examen somatique était sans particularité. L’examen histopathologique d’un prélèvement biopsique de la main avait montré un épiderme, siège d’une hyperkératose orthokératosique avec une colonne verticale de cornéocytes parakératosiques tassés « en pile d’assiettes » (figure 1). Figure 1. Aspect histologique typique de cornéocytes parakératosiques tassés « en pile d’assiette » de la porokératose (GX10, HES). Un infiltrat lymphohistiocytaire occupait le derme superficiel. Cet aspect confirmait le diagnostic de porokératose de Mibelli. Un traitement kératolytique et émollient a été instauré. Commentaire La porokératose de Mibelli (PK) est une dermatose chronique et rare, transmise souvent selon le mode autosomique dominant, à pénétrance variable, apparaissant chez des sujets indemnes de toute maladie (2), mais elle se voit actuellement de plus en plus chez des patients immunodéprimés, notamment les greffés d’organe (3). Caractérisée cliniquement par une bordure kératosique filiforme, parcourue d’un fin sillon en « chemin de ronde » et par une lamelle cornoïde à l’histologie. Formes cliniques Plusieurs formes cliniques de PK ont été décrites : La forme classique représente un peu plus du tiers de tous les cas de PK. Elle se caractérise par une ou plusieurs plaques de taille variable se développant généralement dans l’enfance, habituellement unilatérales et plus rarement bilatérales et symétriques. Cependant, des formes géantes, à bordure surélevée ont été décrites. Les lésions prédominent aux membres, peuvent siéger au visage, mais touchent exceptionnellement les muqueuses et les hémi-muqueuses (4). La PK actinique superficielle disséminée représente la forme la plus fréquente, rapportée le plus souvent dans des pays à fort ensoleillement. Elle touche surtout l’adulte entre la troisième et la quatrième décennie et se manifeste par de nombreuses lésions annulaires de petite taille qui apparaissent de façon bilatérale et symétrique sur des zones photo-exposées (5). La PK superficielle disséminée est cliniquement superposable à la PK actinique superficielle disséminée, mais les lésions siègent indifféremment sur des zones photo-exposées et non exposées (6). La PK linéaire. Les lésions sont unilatérales, systématisées, s’étendant sur un membre selon les lignes de Blaschko, ressemblant à un hamartome épidermique, un psoriasis linéaire ou un lichen striatus. Sur le tronc, les lésions prennent une disposition zostériforme. Cette forme se développe généralement plus tôt dans la vie que les autres formes de PK (7), ce qui était le cas chez notre patient. La PK palmaire et plantaire disséminée se caractérise par des papules kératosiques des paumes et des plantes bilatérales et symétriques apparaissant à l’adolescence. La PK punctata se caractérise par un aspect de petits grains de semoule posés sur la peau, essentiellement au niveau des paumes des mains et des plantes des pieds. Elle se différencie des précédentes par la disparition de la lamelle cornoïde. Les différentes formes peuvent être associées chez le même malade, ou chez des membres d’une même famille, ce qui plaide en faveur de l’unicité des formes de PK (8). L’originalité de notre observation réside dans l’association de deux formes de PK, en plaque et linéaire. Diagnostic Le diagnostic des différentes formes de PK repose sur l’aspect clinique des lésions et les données de l’anamnèse (2). La lésion élémentaire est une papule kératosique initiale à disposition concentrique, qui évolue vers une lésion ovalaire comportant une zone centrale pouvant être lisse, atrophique, hyper- ou hypopigmentée, squameuse ou verruqueuse, et une zone périphérique hyperkératosique. La topographie est variable selon les formes cliniques, le plus souvent bilatérale. Un prurit est présent dans 30 % des cas (5). L’examen histopathologique d’une biopsie faite en bordure d’une lésion montre une colonne verticale de cornéocytes parakératosiques empilés (la lamelle cornoïde), reposant sur une dépression de l’épiderme. Cet aspect, bien que pathognomonique n’est pas spécifique des PK et peut être observé dans les carcinomes basocellulaires, les carcinomes épidermoïdes in situ, les kératoses actiniques, les kératoses séborrhéïques, et certaines verrues (6). Évolution L’évolution est le plus souvent chronique, lente et extensive, par augmentation du nombre et de la taille des lésions. Les PK associées à l’immunosuppression peuvent fluctuer parallèlement au niveau de celle-ci, et des guérisons complètes ont été rapportées après arrêt du traitement immunosuppresseur (9). La transformation maligne des lésions de PK en carcinome épidermoïde, maladie de Bowen, et plus rarement en carcinome basocellulaire survient dans moins de 10 % des cas, après un délai moyen de 30 à 40 ans (10). Pour certains auteurs, les radiations ultraviolettes, l’immunosuppression et la radiothérapie seraient des facteurs favorisant la survenue des lésions de PK, mais également leur dégénérescence (11). Les lésions les plus à risque sont les lésions anciennes de grande taille, siégeant sur les membres, ainsi que les lésions de PK linéaire (12), ce qui est le cas chez notre patient. Les carcinomes sont habituellement uniques, mais peuvent être multiples, avec possibilité de métastases ganglionnaires et d’évolution fatale (10,12). De ce fait, les PK sont souvent considérées comme des dermatoses précancéreuses. Traitement Le traitement de la PK reste peu codifié. L’efficacité des différentes thérapeutiques reste aléatoire et transitoire, plus palliative que curative, visant à réduire le risque de transformation maligne (6). Les kératolytiques, les émollients et les dermocorticoïdes peuvent être utilisés pour des lésions peu étendues (4). L’excision chirurgicale est indiquée pour les lésions de petite taille. Plusieurs essais thérapeutiques utilisant la cryothérapie, le laser CO2 et la dermabrasion ont été rapportés avec des résultats encourageants (6). Le pronostic est globalement favorable, conditionné par le risque de transformation carcinomateuse. Ce risque impose une surveillance régulière, 2 fois par an, accompagnée de conseils de photoprotection, afin de déceler précocement une éventuelle transformation cancéreuse (2). Conclusion La porokératose (PK) de Mibelli est une génodermatose peu commune, d’étiologie inconnue, transmise selon le mode autosomique dominant, se manifestant sous diverses formes. Caractérisée par son aspect clinique fait de lésions atrophiques ou hypertrophiques, bien limitées par un bourrelet filiforme kératosique, et par son aspect histologique (lamelle cornoïde parakératosique). Son pronostic est globalement favorable, mais reste conditionné par la transformation maligne des lésions cutanées qui survient dans moins de 10 % des cas.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :