Publié le 18 sep 2011Lecture 5 min
Le syndrome de Gorlin-Goltz : un cas sporadique
I. KHOUDRI, B. HASSAM, M. AITOURHOUI, Service de dermatologie, CHU Ibn Sina, Rabat, Maroc
Le syndrome de Gorlin-Goltz encore appelé nævomatose basocellulaire, phacomatose de Gorlin-Goltz, ou cinquième phacomatose, est une génodermatose rare transmise selon le mode autosomique dominant (1). Nous présentons ici un nouveau cas de syndrome de Gorlin-Goltz particulier par son caractère sporadique. Plus de 600 cas de syndrome de Gorlin-Goltz sont rapportés dans la littérature. Il s’agit d’une dysembryoplasie impliquant les systèmes d’origine ectodermique et mésodermiques. Le syndrome de Gorlin-Goltz se caractérise par des anomalies de développement et une prédisposition à différents cancers (2). Il associe sur le plan clinique des nævi et des carcinomes basocellulaires faciaux, des kystes épidermoïdes des maxillaires à des malformations squelettiques (3).
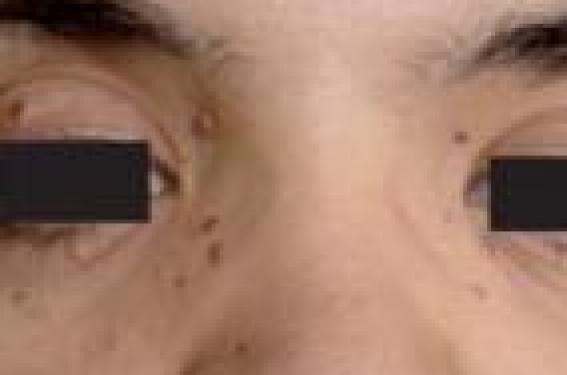
Observation E. N., âgée de 29 ans, issue d’un mariage non consanguin, sans antécédent familial, opérée à l’âge de 16 ans pour une scoliose malformative cervico-dorsale, a consulté pour des lésions papuleuses pigmentées bilatérales périorbitaires (figure 1) apparues dès l’âge de 5 ans. Figure 1. Nævi nævo-cellulaires multiples périorbitaires, et hypertélorisme. Ces lésions sont devenues prurigineuses, avec apparition il y a 2 ans d’autres lésions pigmentées éparpillées au niveau du visage, le cou et le tronc (figure 2). L’examen clinique retrouve plusieurs nævi bilatéraux périorbitaires de taille inférieure à 0,5 cm, associés à plusieurs lésions arrondies, pigmentées, à contours perlés, infiltrées par endroits, de tailles variables, allant de 0,3 à 1 cm de diamètre, éparpillées au niveau de la région temporale droite, au niveau du cou, la région lombaire et de l’abdomen (figure 2). Certaines lésions sont évocatrices de carcinomes basocellulaires. L’examen dermatologique retrouve également des « pits » (puits de kératine) palmaires. L’examen ostéoarticulaire révèle des anomalies squelettiques à type de : palais ogival, hypertélorisme, protrusion sternale, cyphose dorsale et déformation de l’omoplate droite. Le reste de l’examen somatique est normal en dehors d’un mauvais état bucco-dentaire. Malgré l’absence de cas similaire dans la famille, un syndrome de Gorlin- Goltz sporadique est évoqué. La biopsie exérèse des lésions cutanées suspectes révèle des nævi nævo-cellulaires sans signes histologiques de carcinome basocellulaire. La radiographie thoracique montre une synostose costale de l’arc moyen de la 4e et 5e côte (figure 3). Au niveau du rachis, une cypho-scoliose cervico-dorsale à concavité droite est notée. La radiographie des extrémités est normale. La radiographie panoramique dentaire révèle des kystes au niveau des maxillaires supérieurs et inférieurs. Le scanner cérébral objective des calcifications de la faux du cerveau. Figure 2. Lésions pigmentées infiltrées à contours perlés du tronc (nævi nævo-cellulaires à l’histologie). L’étude du matériel génétique n’a pas été réalisée. L’enquête familiale complétée par un examen clinique ne révèle pas de cas similaire. Sur le plan thérapeutique, la patiente a été traitée par électrocoagulation avec curetage pour certaines lésions, et par exérèse chirurgicale pour d’autres. Une photoprotection externe a été également recommandée, ainsi qu’une surveillance régulière des lésions cutanées. Discussion Le syndrome de Gorlin-Goltz a été individualisé pour la première fois en 1960 par un stomatologiste, R.J. Gorlin et un dermatologiste, R.W. Goltz (4). Sa prévalence est estimée entre 1/57 000 et 1/164 000 (5). C’est une dysembryoplasie qui se transmet selon un mode autosomique dominant avec habituellement une forte pénétrance (97 %) et une expression variable(6). Notre observation se caractérise par l’absence du caractère familial et héréditaire de cette affection. Figure 3. Radiographie du thorax. Synostose costale (arc moyen de la 4e-5e côte), scoliose vertébrale, matériel d’ostéosynthèse scapulaire. L’étude génétique n’a pas été réalisée dans notre cas, cependant des progrès récents ont montré que cette maladie résulte d’une mutation ponctuelle d’un gène tumoro-suppresseur appelé PTCH. Il s’agit d’un gène de développement localisé sur le chromosome 9q22.3-31 (1,7). Aspects cliniques Cette pathologie s’exprime par des manifestations cliniques très variées (2). Les signes cutanés sont dominés par les nævi basocellulaires (99 %) à localisation centro-faciale surtout ou thoracique. Ces nævi sont dotés d’un grand potentiel évolutif de dégénérescence maligne en carcinomes basocellulaires. Les « pits » palmo-plantaires (16,7 %) sous forme de petites dépressions épidermiques punctiformes sont très caractéristiques. Les signes osseux sont dominés par les kystes osseux maxillaires (75 %) dépistés radiologiquement ou découverts à l’occasion d’accidents infectieux ou d’une anomalie dentaire. Les anomalies squelettiques touchent essentiellement les côtes (synostose, bifidité, agénésie), la colonne vertébrale (cypho-scoliose, spina bifida) et les mains (bradymétacarpisme). Les anomalies neurologiques sont surtout à type de calcification intracrânienne (93 %). D’autres manifestations ont été décrites telles qu’un glaucome, cataracte, hypogonadisme (6). Le diagnostic positif repose sur la présence de deux critères majeurs ou l’association d’un critère majeur et deux critères mineurs (tableau) (5). Le diagnostic du syndrome de Gorlin-Goltz a été retenu chez notre patiente sur deux critères majeurs que sont : les « pits » palmaires et les calcifications intracrâniennes. D’autres signes étaient en faveur du diagnostic, à savoir les anomalies costales et l’hypertélorisme. L’étude histologique n’a pas été réalisée pour les kystes maxillaires, et aucun carcinome basocellulaire n’a été retrouvé à l’étude anatomopathologique cutanée. Par ailleurs, notre observation semble être un cas sporadique vu l’absence de cas similaires dans la famille. Il s’agit du troisième cas colligé dans notre service avec un intervalle de 6 et 4 ans ce qui suggère la rareté de cette pathologie dans notre contexte. L’intérêt de cette observation est de rappeler également la nécessité du dépistage précoce car il s’agit d’une pathologie chronique prédisposant à certaines néoplasies (carcinomes basocellulaires, médulloblastomes). Des mesures de photoprotection ainsi qu’une surveillance régulière s’imposent pour éviter ces complications. Sans oublier l’intérêt du conseil génétique. Conclusion Le syndrome de Gorlin-Goltz est une entité clinique rare et très polymorphe pouvant nécessiter une approche multidisciplinaire. Les manifestations dermatologiques sont au premier rang de cette affection. Le caractère autosomique dominant de cette affection n’est pas toujours retrouvé. Un diagnostic précoce et une surveillance périodique sont nécessaires pour éviter les complications.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



")